

Decontamination of ambient RNA in single-cell RNA-seq with DecontX
- PMID: 32138770
- PMCID: PMC7059395
- DOI: 10.1186/s13059-020-1950-6
Decontamination of ambient RNA in single-cell RNA-seq with DecontX
Abstract
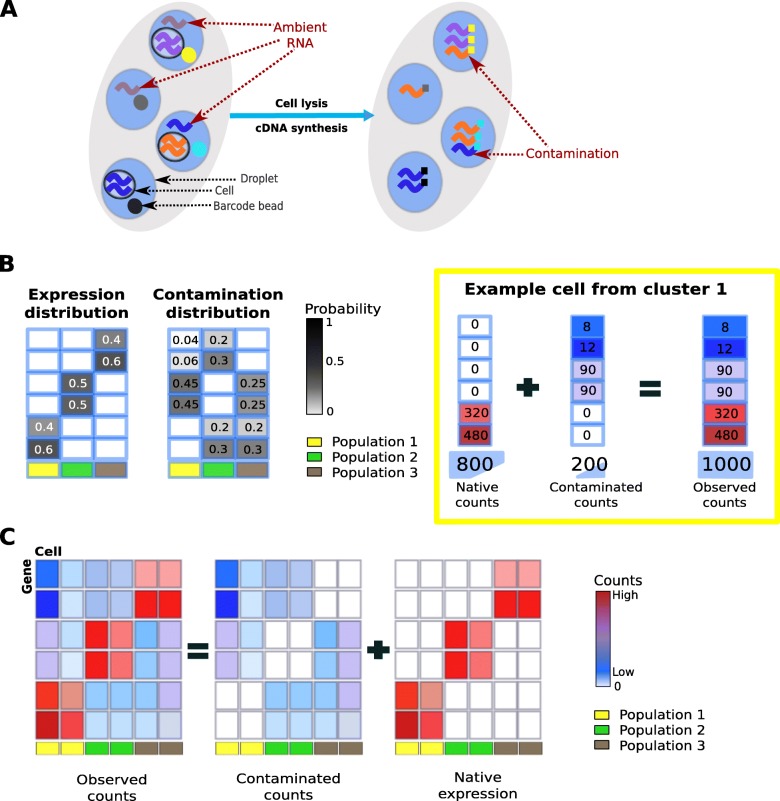
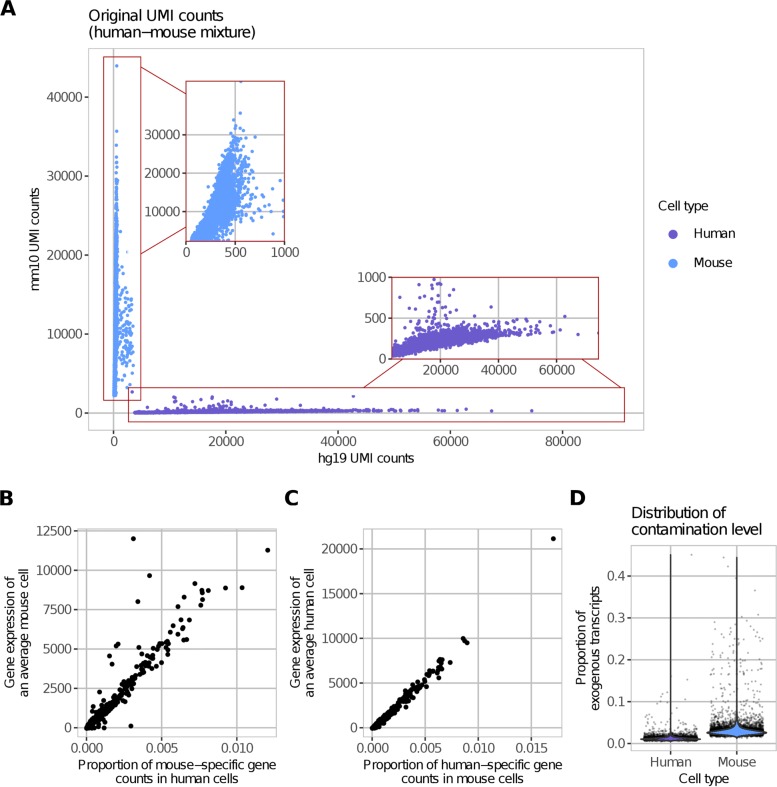
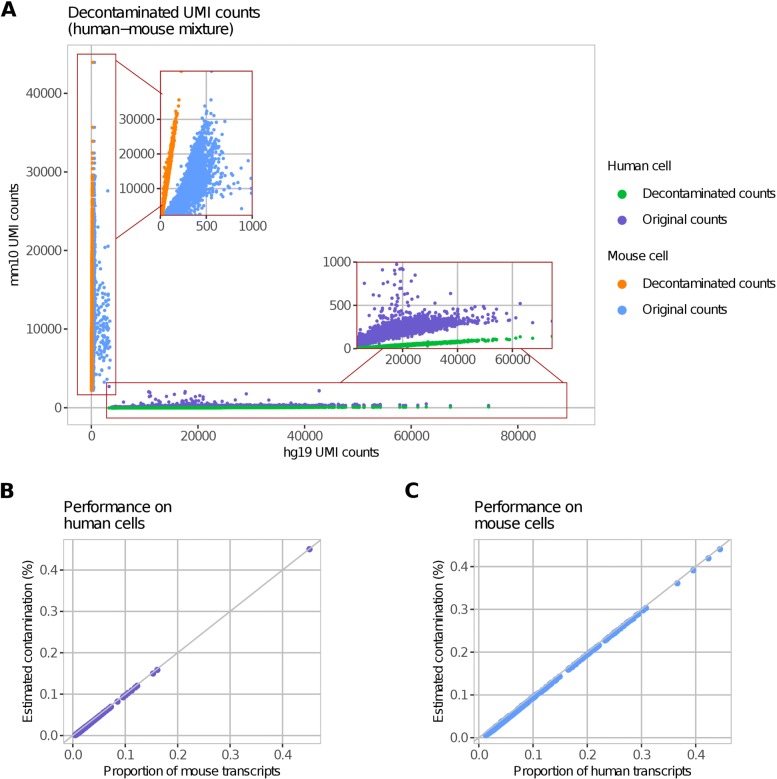
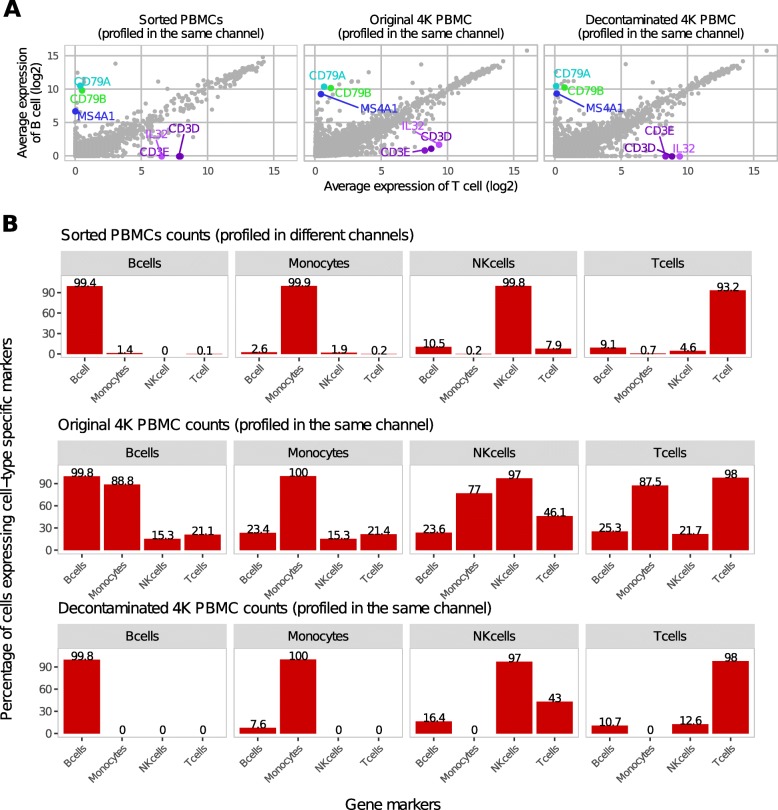
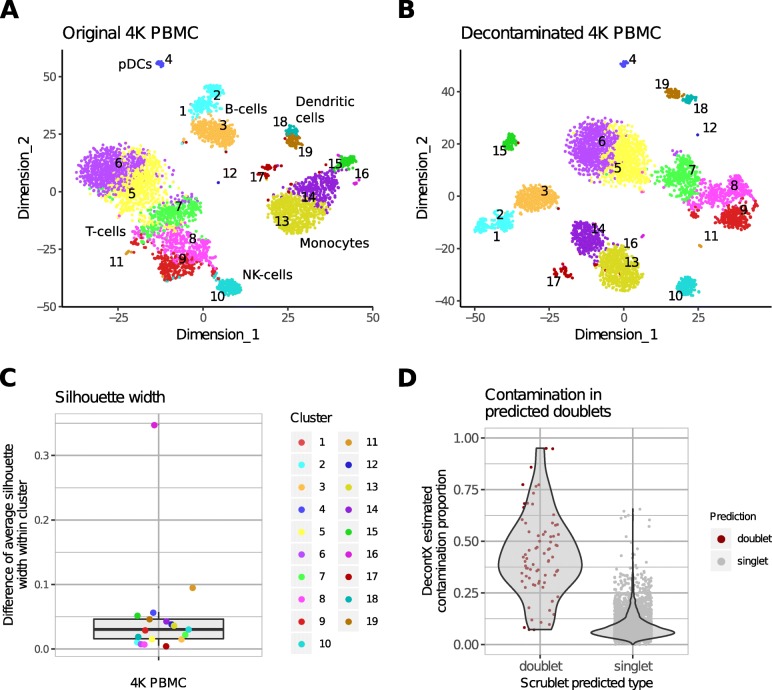
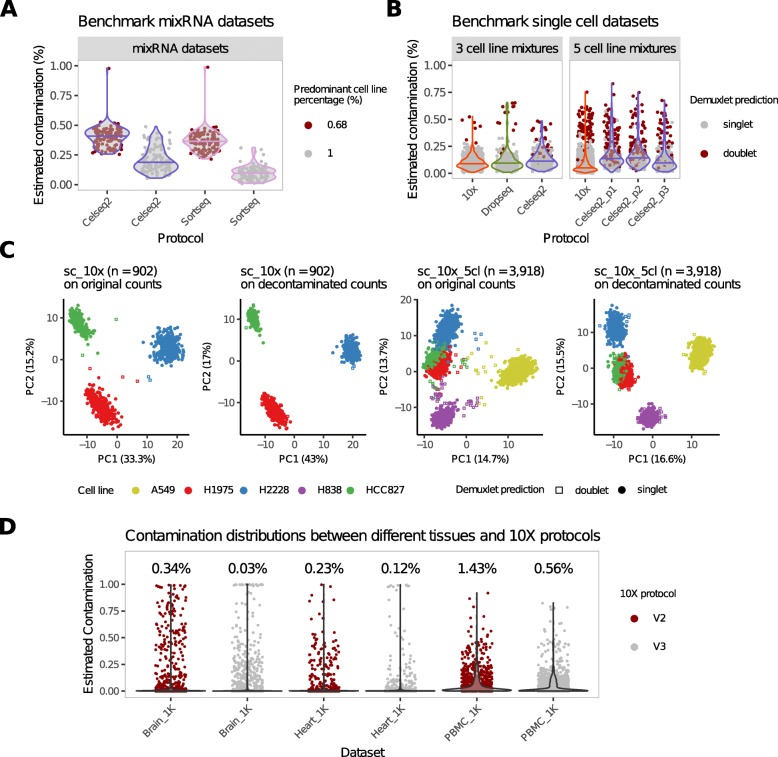
Droplet-based microfluidic devices have become widely used to perform single-cell RNA sequencing (scRNA-seq). However, ambient RNA present in the cell suspension can be aberrantly counted along with a cell's native mRNA and result in cross-contamination of transcripts between different cell populations. DecontX is a novel Bayesian method to estimate and remove contamination in individual cells. DecontX accurately predicts contamination levels in a mouse-human mixture dataset and removes aberrant expression of marker genes in PBMC datasets. We also compare the contamination levels between four different scRNA-seq protocols. Overall, DecontX can be incorporated into scRNA-seq workflows to improve downstream analyses.
Keywords: Bayesian mixture model; Decontamination; Single cell; scRNA-seq.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
