

Glanzmann thrombasthenia: genetic basis and clinical correlates
- PMID: 32139434
- PMCID: PMC7109743
- DOI: 10.3324/haematol.2018.214239
Glanzmann thrombasthenia: genetic basis and clinical correlates
Abstract
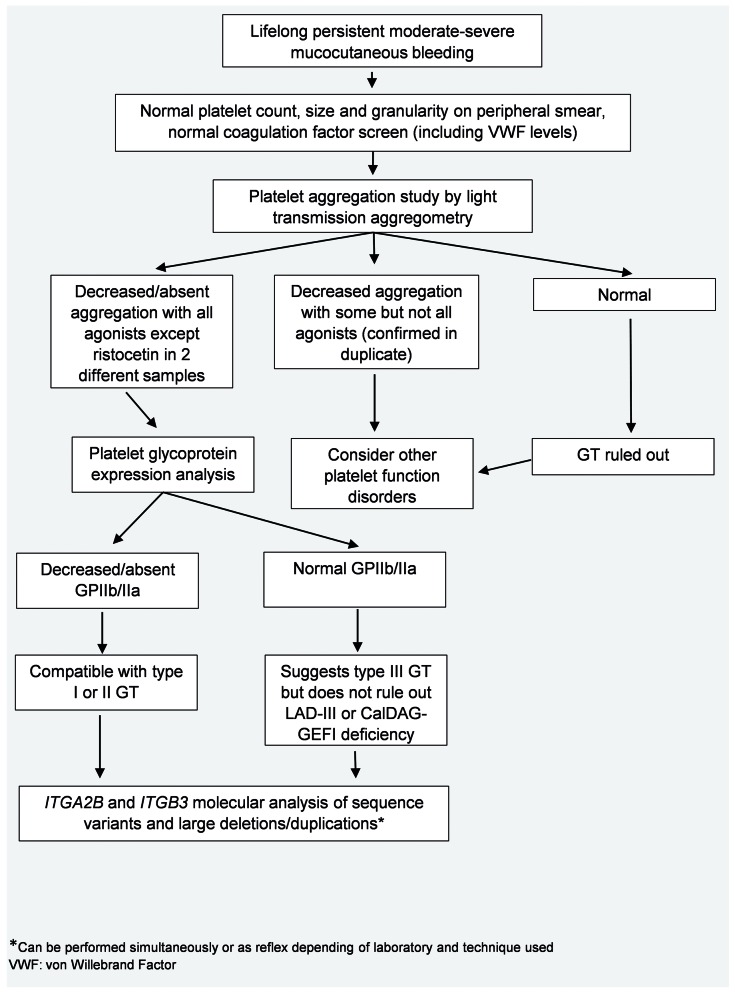
Glanzmann thrombasthenia (GT) is an autosomal recessive disorder of platelet aggregation caused by quantitative or qualitative defects in integrins αIIb and β3. These integrins are encoded by the ITGA2B and ITGB3 genes and form platelet glycoprotein (GP)IIb/IIIa, which acts as the principal platelet receptor for fibrinogen. Although there is variability in the clinical phenotype, most patients present with severe mucocutaneous bleeding at an early age. A classic pattern of abnormal platelet aggregation, platelet glycoprotein expression and molecular studies confirm the diagnosis. Management of bleeding is based on a combination of hemostatic agents including recombinant activated factor VII with or without platelet transfusions and antifibrinolytic agents. Refractory bleeding and platelet alloimmunization are common complications. In addition, pregnant patients pose unique management challenges. This review highlights clinical and molecular aspects in the approach to patients with GT, with particular emphasis on the significance of multidisciplinary care.
Copyright© 2020 Ferrata Storti Foundation.
Figures

References
-
- Commentary on and reprint of Glanzmann E, Hereditäre häemorrhagische thrombasthenie. Ein Beitrag zur Pathologie der Blutplättchen [Hereditary hemorrhagic thrombasthenia: A contribution on the pathology of blood platelets], in Jahrbuch für Kinderheilkunde (1918) 88:113–141. In: Lichtman MA, Spivak JL, Boxer LA, Shattil SJ, Henderson ES, eds. Hematology. San Diego: Academic Press, 2000:55–III.
-
- Caen J, Cousin C. [“In vivo” disorder of platelet adhesiveness in Willebrand’s disease and Glanzmann’s thrombasthenias. Trial interpretation]. Nouv Rev Fr Hematol. 1962;2:685–694. - PubMed
-
- Caen JP, Castaldi PA, Leclerc JC, et al. Congenital bleeding disorders with long bleeding time and normal platelet count: I. Glanzmann’s thrombasthenia (report of fifteen patients). Am J Med. 1966;41(1):4–26.
-
- Nurden AT, Caen JP. An abnormal platelet glycoprotein pattern in three cases of Glanzmann’s thrombasthenia. Br J Haematol. 1974;28(2):253–260. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
