

Genetic and phenotypic landscape of the mitochondrial genome in the Japanese population
- PMID: 32139841
- PMCID: PMC7058612
- DOI: 10.1038/s42003-020-0812-9
Genetic and phenotypic landscape of the mitochondrial genome in the Japanese population
Abstract
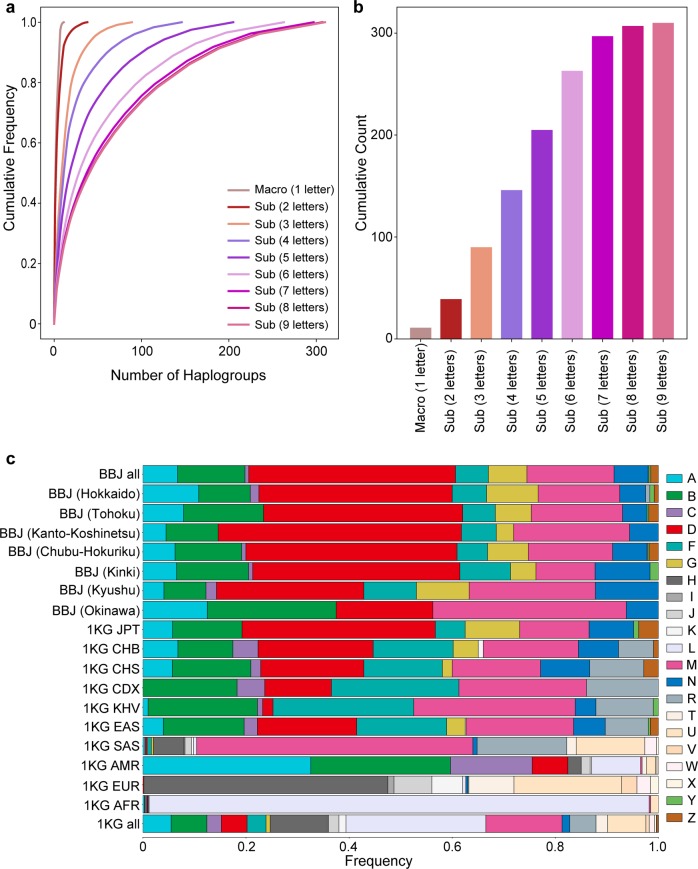
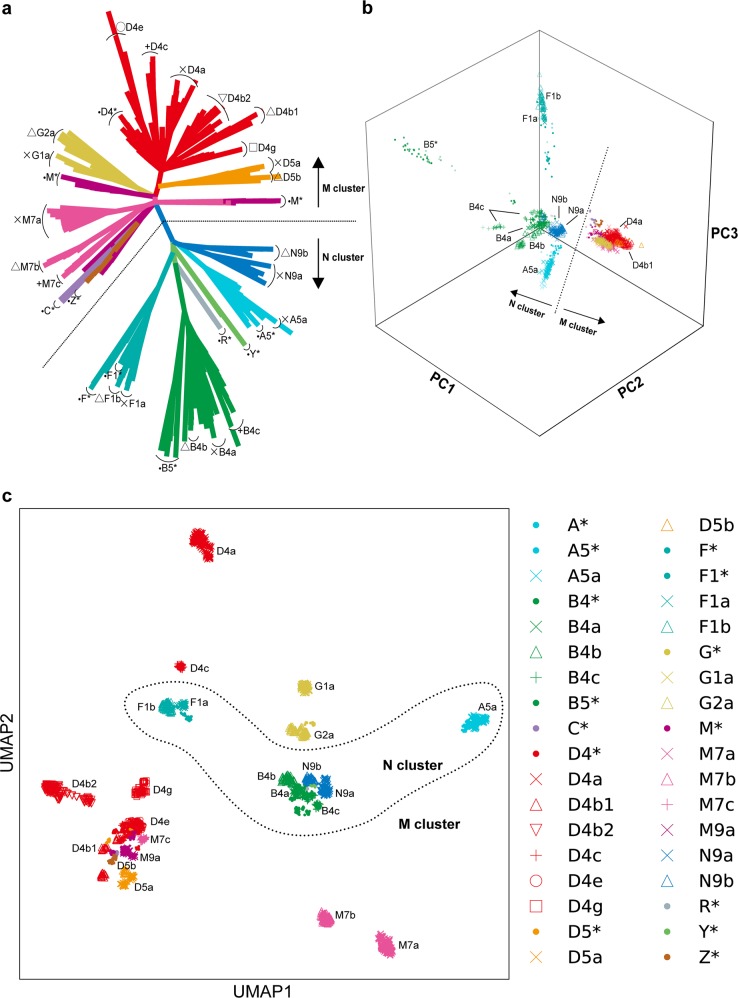
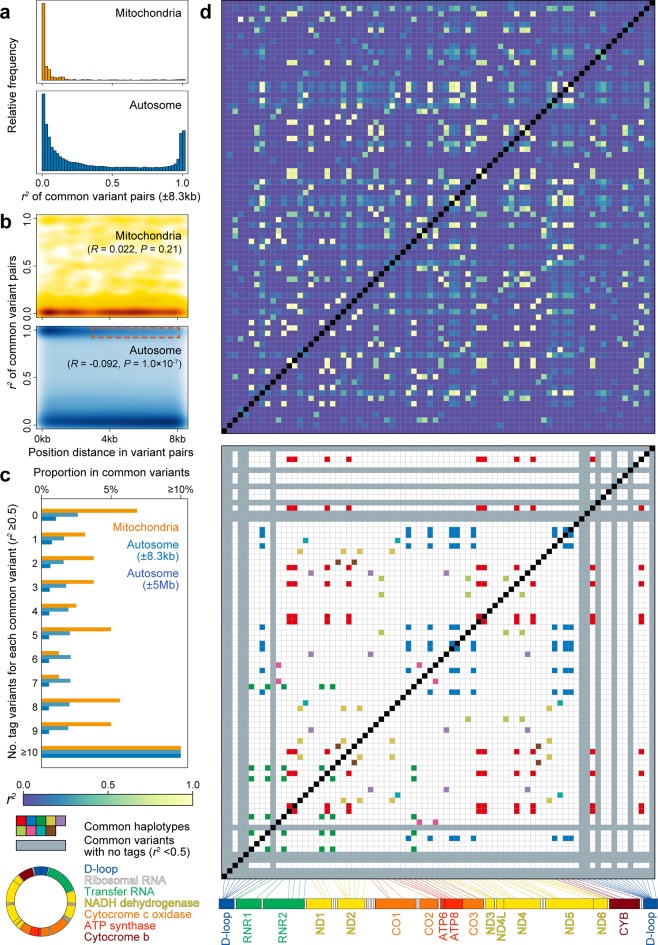
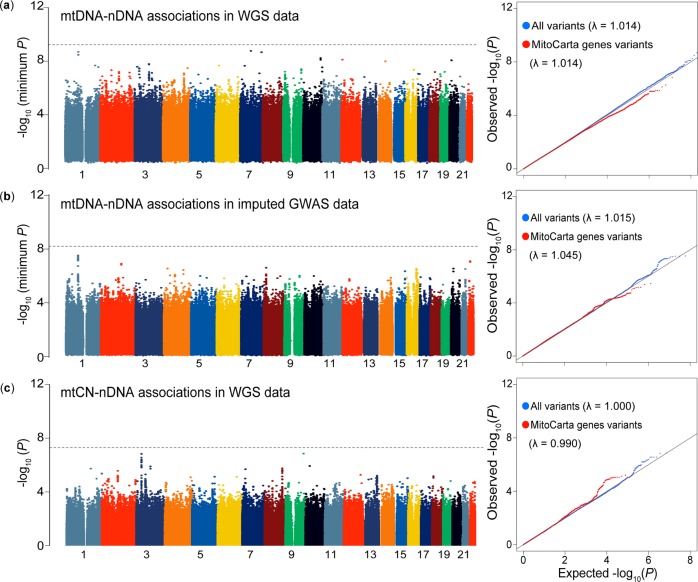
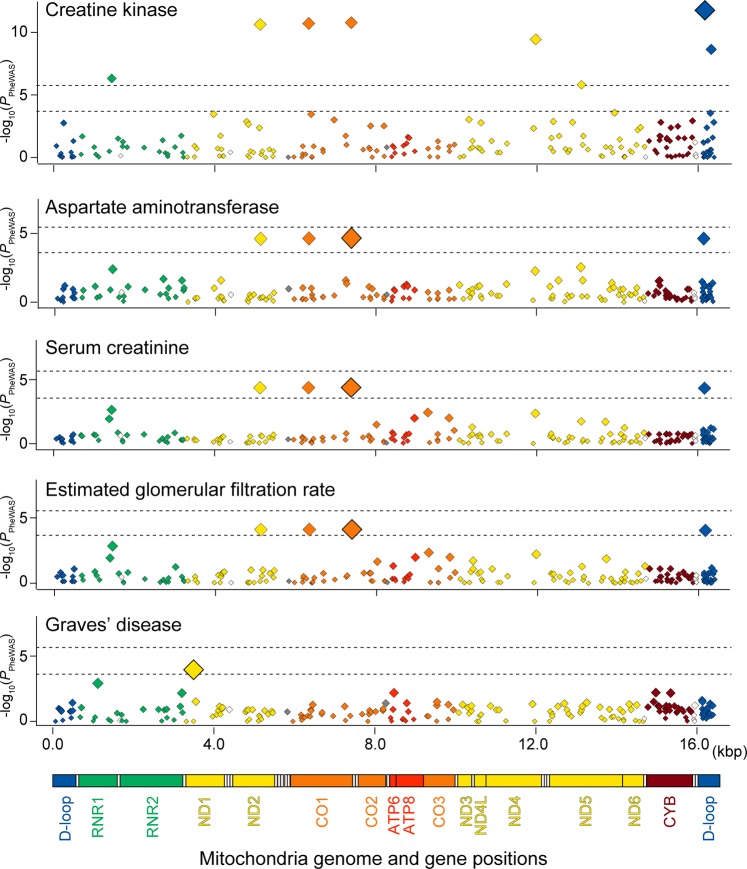
The genetic landscape of mitochondrial DNA (mtDNA) has been elusive. By analyzing mtDNA using the whole genome sequence (WGS) of Japanese individuals (n = 1928), we identified 2023 mtDNA variants and high-resolution haplogroups. Frequency spectra of the haplogroups were population-specific and were heterogeneous among geographic regions within Japan. Application of machine learning methods could finely classify the subjects corresponding to the high-digit mtDNA sub-haplogroups. mtDNA had distinct genetic structures from that of nuclear DNA (nDNA), characterized by no distance-dependent linkage disequilibrium decay, sparse tagging of common variants, and the existence of common haplotypes spanning the entire mtDNA. We did not detect any evidence of mtDNA-nDNA (or mtDNA copy number-nDNA) genotype associations. Together with WGS-based mtDNA variant imputation, we conducted a phenome-wide association study of 147,437 Japanese individuals with 99 clinical phenotypes. We observed pleiotropy of mtDNA genetic risk on the five late-onset human complex traits including creatine kinase (P = 1.7 × 10-12).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
