

Interplay between whole-genome doubling and the accumulation of deleterious alterations in cancer evolution
- PMID: 32139907
- PMCID: PMC7116784
- DOI: 10.1038/s41588-020-0584-7
Interplay between whole-genome doubling and the accumulation of deleterious alterations in cancer evolution
Abstract
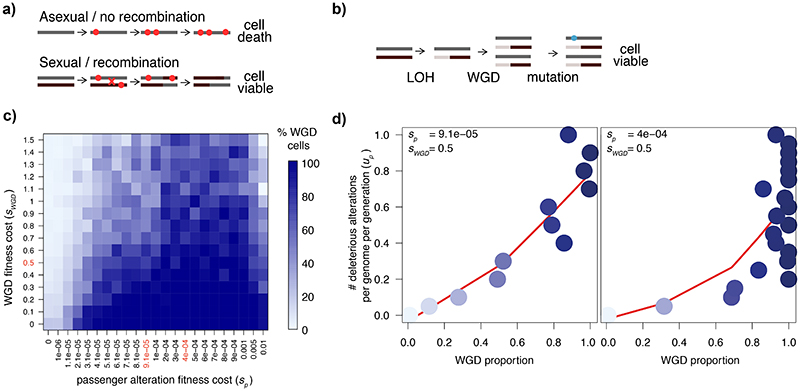
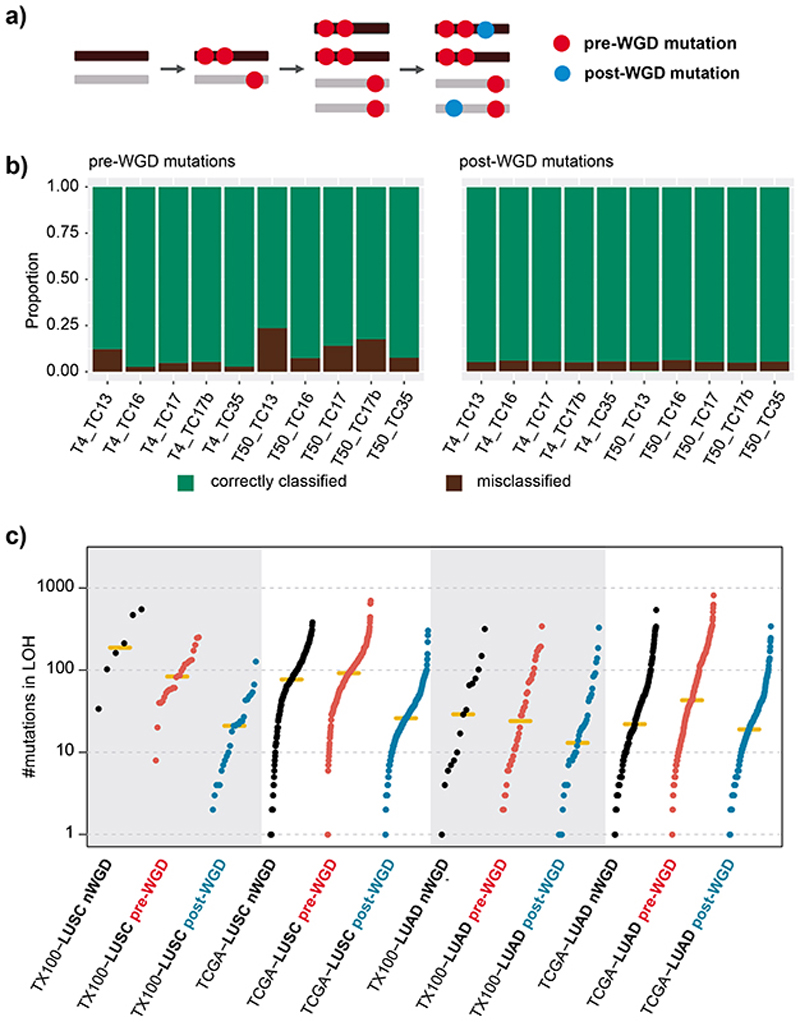
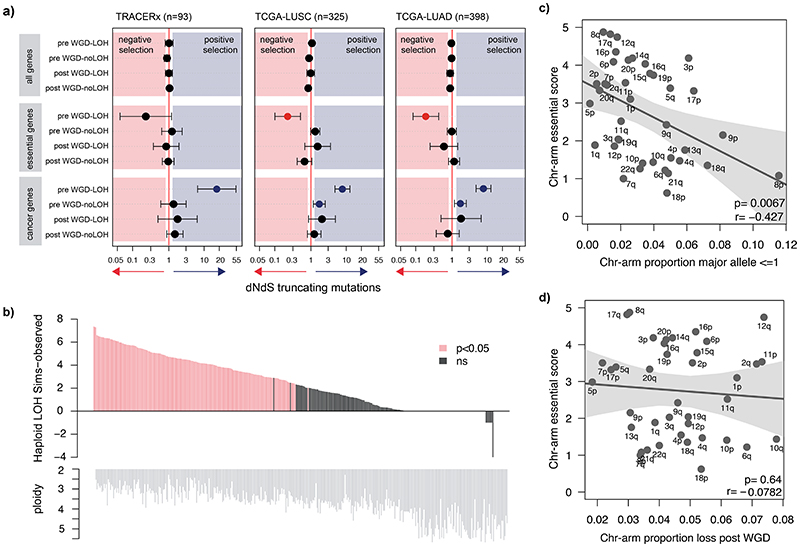
Whole-genome doubling (WGD) is a prevalent event in cancer, involving a doubling of the entire chromosome complement. However, despite its prevalence and prognostic relevance, the evolutionary selection pressures for WGD in cancer have not been investigated. Here, we combine evolutionary simulations with an analysis of cancer sequencing data to explore WGD during cancer evolution. Simulations suggest that WGD can be selected to mitigate the irreversible, ratchet-like, accumulation of deleterious somatic alterations, provided that they occur at a sufficiently high rate. Consistent with this, we observe an enrichment for WGD in tumor types with extensive loss of heterozygosity, including lung squamous cell carcinoma and triple-negative breast cancers, and we find evidence for negative selection against homozygous loss of essential genes before, but not after, WGD. Finally, we demonstrate that loss of heterozygosity and temporal dissection of mutations can be exploited to identify novel tumor suppressor genes and to obtain a deeper characterization of known cancer genes.
Conflict of interest statement
The authors declare competing financial interests: C.S. receives grant support from Pfizer, AstraZeneca, BMS, and Ventana. C.S. has consulted for Boehringer Ingelheim, Eli Lily, Servier, Novartis, Roche-Genentech, GlaxoSmithKline, Pfizer, BMS, Celgene, AstraZeneca, Illumina, and Sarah Cannon Research Institute. C.S. is a shareholder of Apogen Biotechnologies, Epic Bioscience, GRAIL, and has stock options and is co-founder of Achilles Therapeutics. N.M. and G.W. has stock options and has consulted for Achilles Therapeutics.
Figures


Comment in
-
Weighing up effects of extra chromosomes.Nat Rev Cancer. 2020 May;20(5):259. doi: 10.1038/s41568-020-0257-y. Nat Rev Cancer. 2020. PMID: 32235903 No abstract available.
References
-
- Storchova Z, Pellman D. From polyploidy to aneuploidy, genome instability and cancer. Nat Rev Mol Cell Biol. 2004;5:45–54. - PubMed
-
- Huxley J. Evolution The modern synthesis. George Alien & Unwin Ltd; London: 1942.
Publication types
MeSH terms
Substances
Grants and funding
- 21999/CRUK_/Cancer Research UK/United Kingdom
- 30025/CRUK_/Cancer Research UK/United Kingdom
- 218274/WT_/Wellcome Trust/United Kingdom
- 211179/Z/18/Z/WT_/Wellcome Trust/United Kingdom
- FC001169/WT_/Wellcome Trust/United Kingdom
- 20465/CRUK_/Cancer Research UK/United Kingdom
- 617844/ERC_/European Research Council/International
- FC001202/WT_/Wellcome Trust/United Kingdom
- 835297/ERC_/European Research Council/International
- FC001169, FC001202/MRC_/Medical Research Council/United Kingdom
- 23896/CRUK_/Cancer Research UK/United Kingdom
- 24956/CRUK_/Cancer Research UK/United Kingdom
- MR/P014712/2/MRC_/Medical Research Council/United Kingdom
- MC_UP_1203/1/MRC_/Medical Research Council/United Kingdom
- 17786/CRUK_/Cancer Research UK/United Kingdom
- 211179/WT_/Wellcome Trust/United Kingdom
- 19278/CRUK_/Cancer Research UK/United Kingdom
- A23896/CRUK_/Cancer Research UK/United Kingdom
- MR/P014712/1/MRC_/Medical Research Council/United Kingdom
- 29420/CRUK_/Cancer Research UK/United Kingdom
- 29569/CRUK_/Cancer Research UK/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
