

TMEM173 Drives Lethal Coagulation in Sepsis
- PMID: 32142632
- PMCID: PMC7316085
- DOI: 10.1016/j.chom.2020.02.004
TMEM173 Drives Lethal Coagulation in Sepsis
Abstract
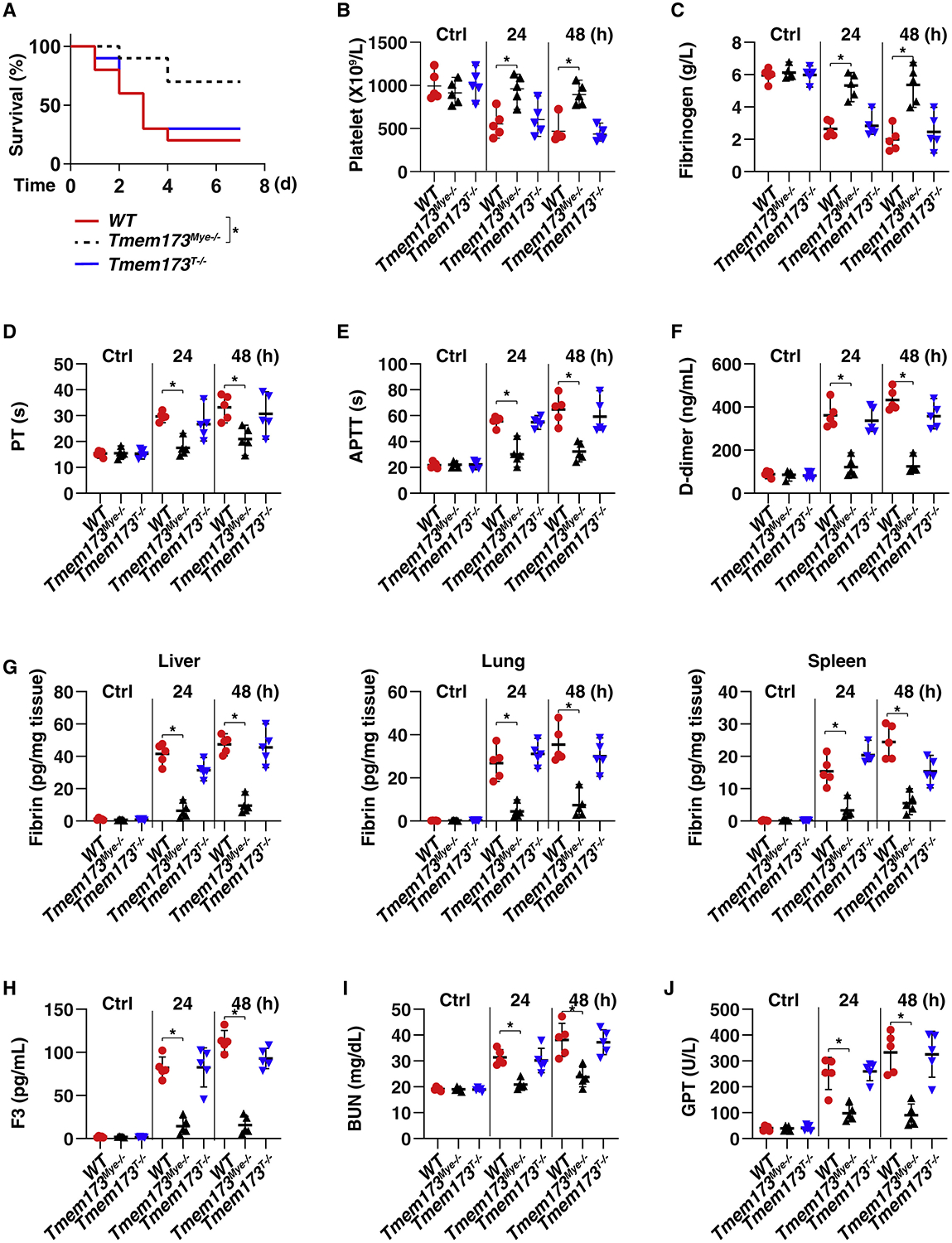
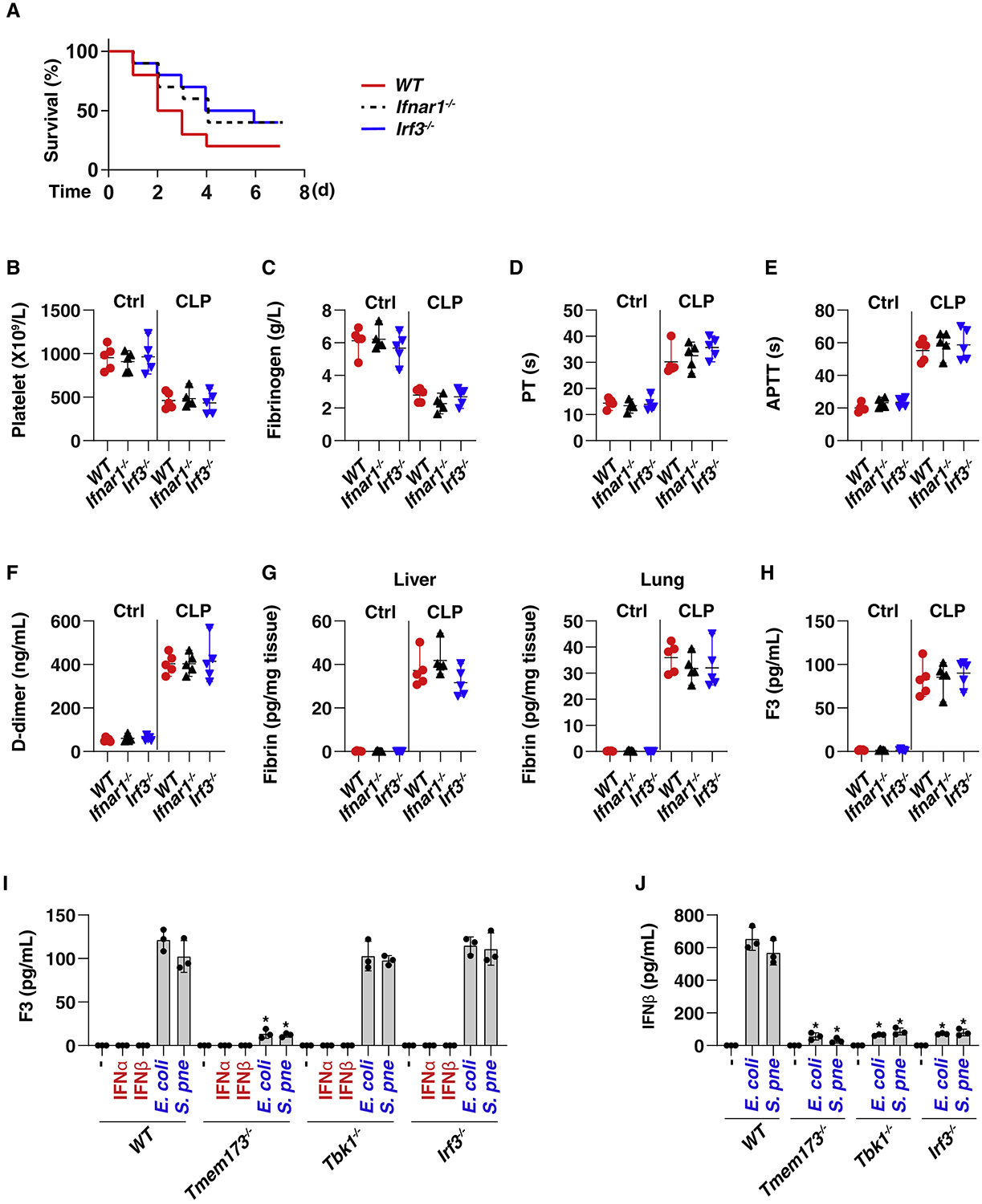
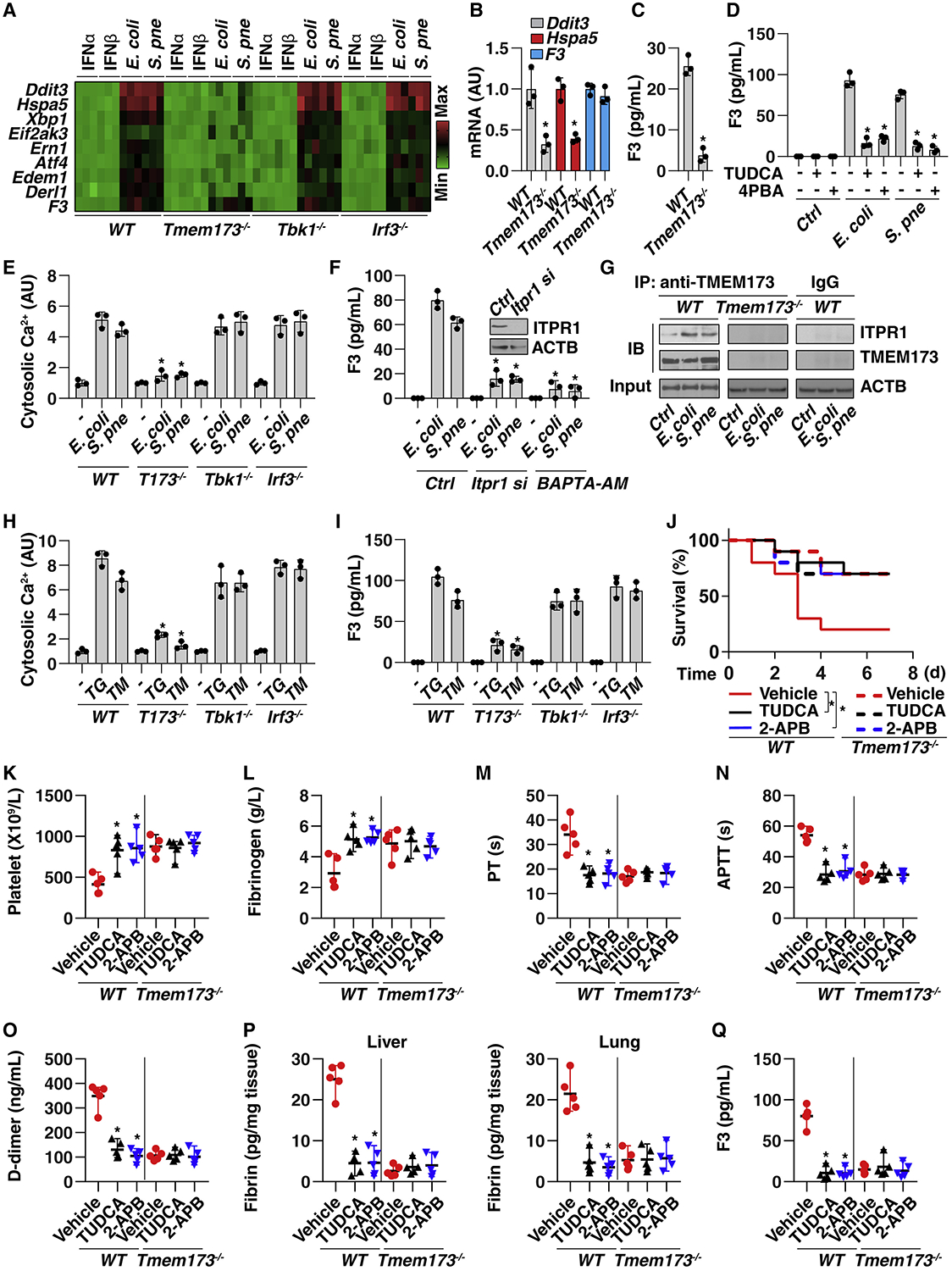
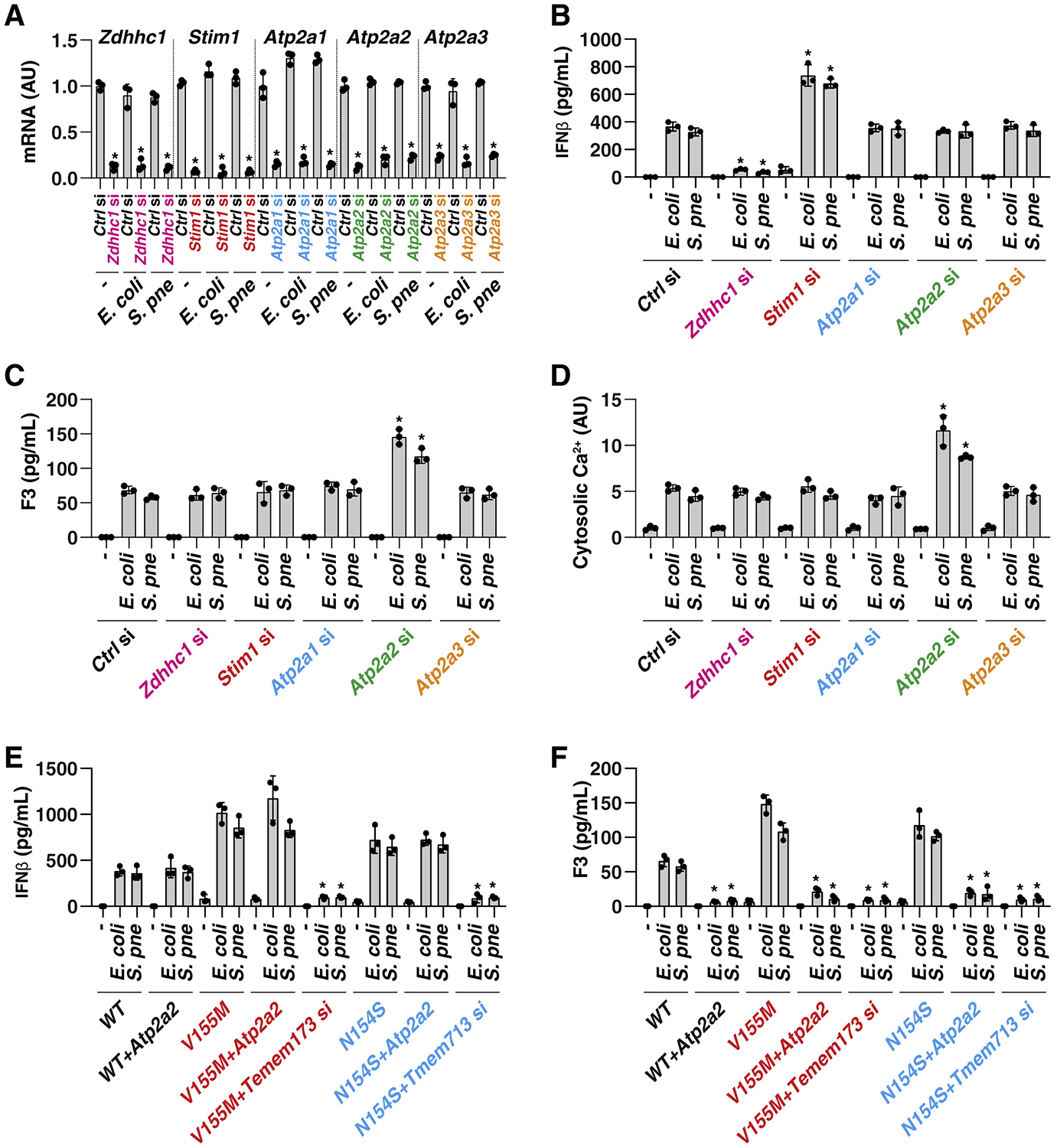
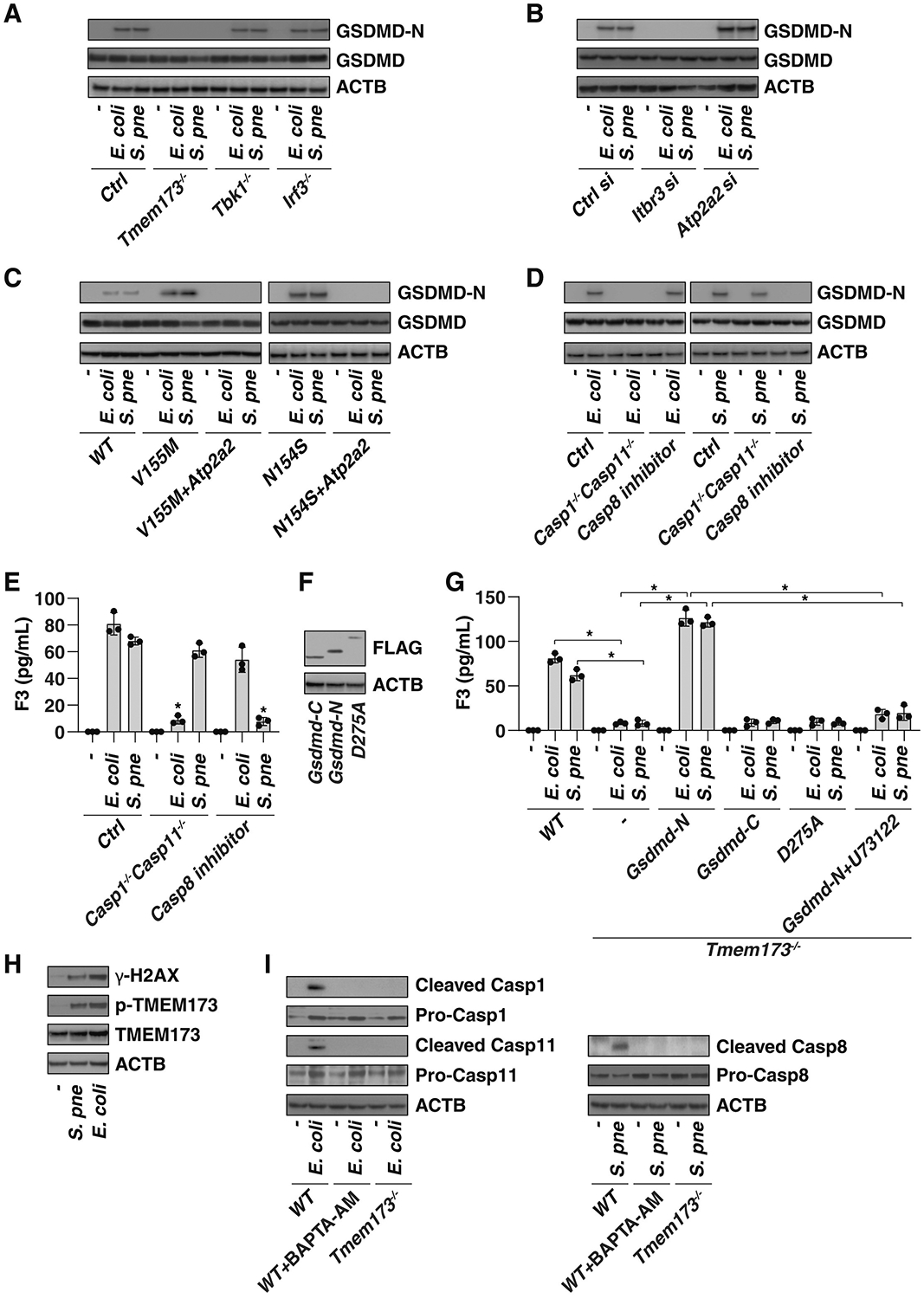
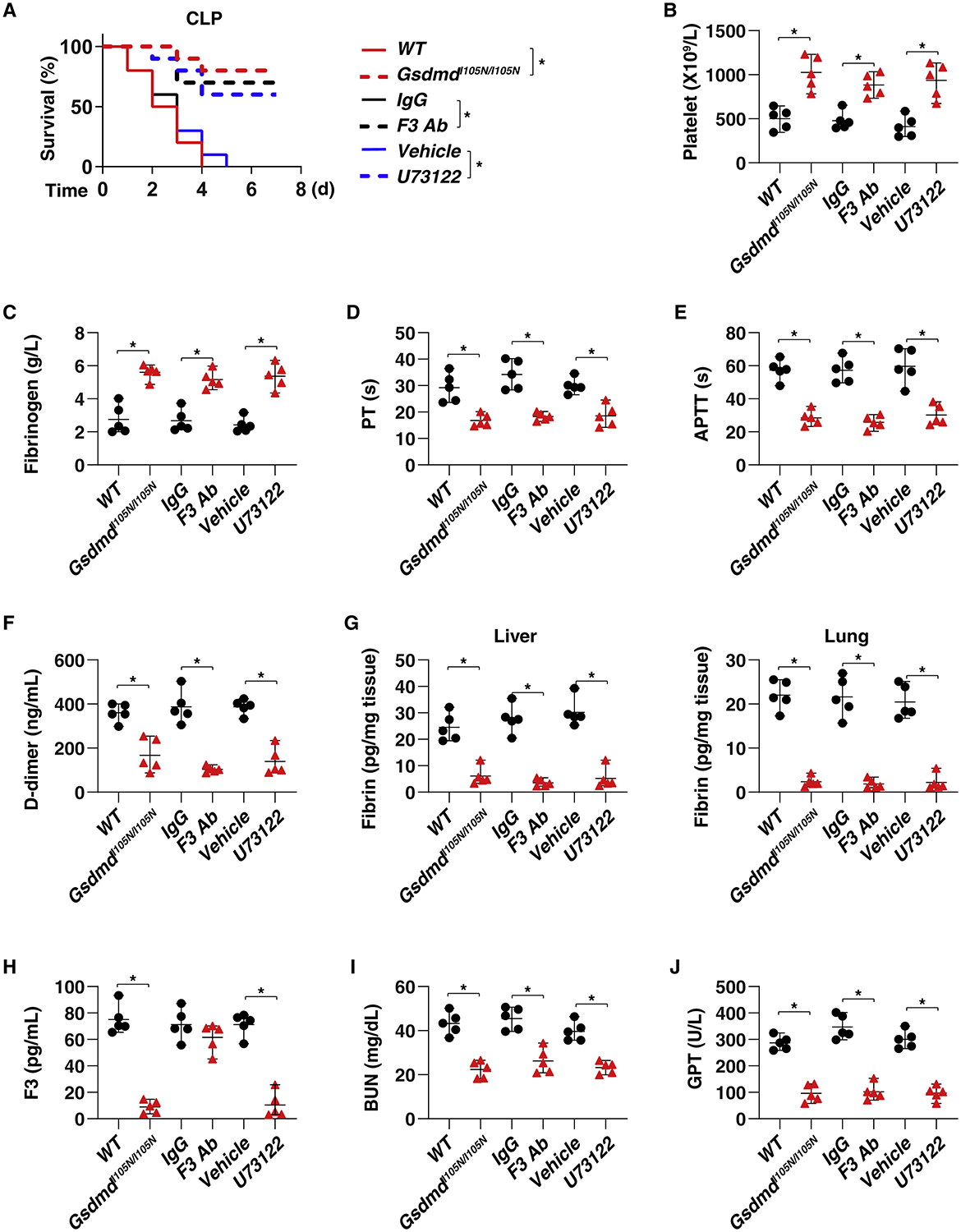
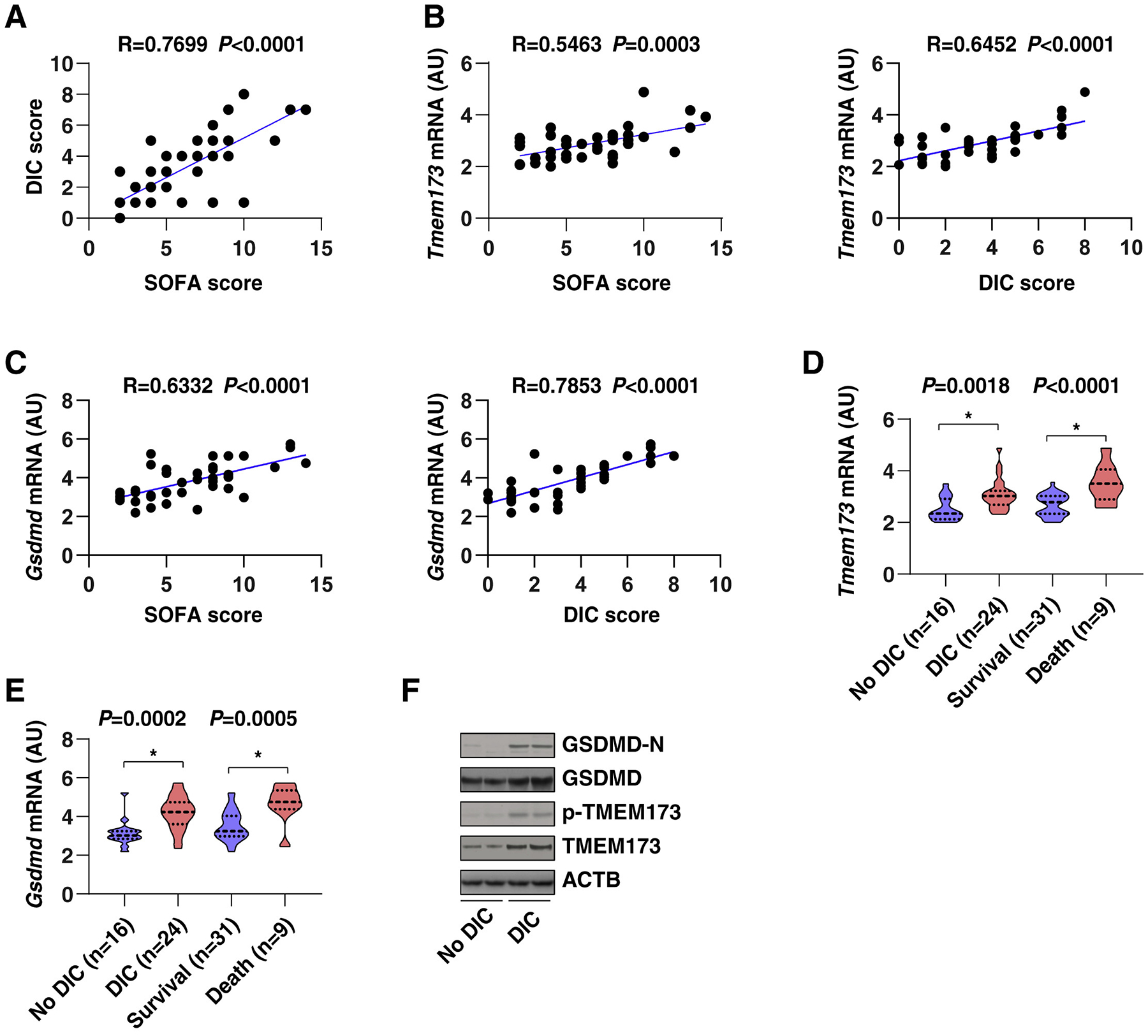
The discovery of TMEM173/STING-dependent innate immunity has recently provided guidance for the prevention and management of inflammatory disorders. Here, we show that myeloid TMEM173 occupies an essential role in regulating coagulation in bacterial infections through a mechanism independent of type I interferon response. Mechanistically, TMEM173 binding to ITPR1 controls calcium release from the endoplasmic reticulum in macrophages and monocytes. The TMEM173-dependent increase in cytosolic calcium drives Gasdermin D (GSDMD) cleavage and activation, which triggers the release of F3, the key initiator of blood coagulation. Genetic or pharmacological inhibition of the TMEM173-GSDMD-F3 pathway blocks systemic coagulation and improves animal survival in three models of sepsis (cecal ligation and puncture or bacteremia with Escherichia coli or Streptococcus pneumoniae infection). The upregulation of the TMEM173 pathway correlates with the severity of disseminated intravascular coagulation and mortality in patients with sepsis. Thus, TMEM173 is a key regulator of blood clotting during lethal bacterial infections.
Keywords: ER stress; GSDMD; STING; TMEM173; calcium; coagulation; inflammasome; pyroptosis; sepsis; tissue factor.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no conflicts of interest or financial interests.
Figures
Comment in
-
Taking the STING Out of Sepsis?Cell Host Microbe. 2020 Apr 8;27(4):491-493. doi: 10.1016/j.chom.2020.03.016. Cell Host Microbe. 2020. PMID: 32272071
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
