

A case report of a novel 22 bp duplication within exon 1 of the UGT1A1 in a Sudanese infant with Crigler-Najjar syndrome type I
- PMID: 32143638
- PMCID: PMC7060512
- DOI: 10.1186/s12876-020-01192-4
A case report of a novel 22 bp duplication within exon 1 of the UGT1A1 in a Sudanese infant with Crigler-Najjar syndrome type I
Abstract
Background: Crigler Najjar type 1 is a rare autosomal recessive condition caused by the absence of UDPGT enzyme due to mutations in the UGT1A1 gene. This enzyme is responsible for elimination of unconjugated bilirubin from the body by glucuronidation. Affected individuals are at risk for kernicterus and require lifelong phototherapy. Liver transplant is the only definitive treatment.
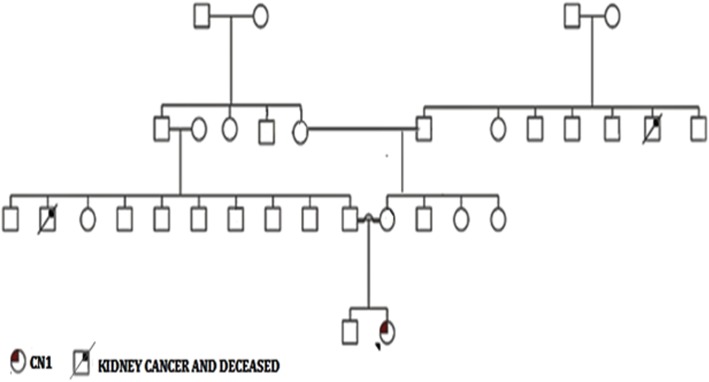
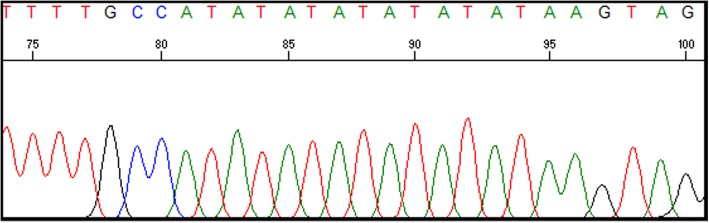
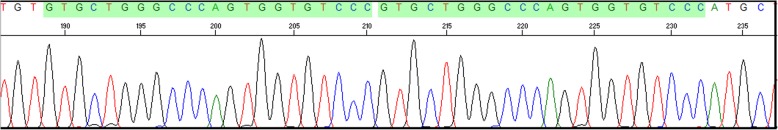
Case presentation: Here we report a case of a 6 month old Sudanese female infant with CN1 whose molecular analysis revealed a novel homozygous 22 base pair duplication (c.55_76dup) in the coding exon 1 of the UGT1A1 gene. This 22 bp duplication causes a frame shift leading to a premature stop codon. She underwent a successful liver transplant at 7 months of age and is doing well at 1 year follow-up.
Conclusion: This study shows that molecular diagnosis helps in precise diagnosis of CN1 and in prognosis, prompt medical intervention and appropriate therapy. This particular 22 bp duplication within the coding region of UGT1A1 can be a founder mutation in the Sudanese population.
Keywords: Autosomal recessive; Bilirubin; Crigler-Najjar syndrome; Liver transplant; Mutation; UGT1A1.
Conflict of interest statement
SV, KKM, DRV,LKVM are employees of GenesNLife Healthcare Pvt., Ltd. CCK, MR and NS are employees of Gleneageles Global hospitals.
Figures
References
-
- Kadakol A, Ghosh SS, Sappal BS, Sharma G, Chowdhury JR, Chowdhury NR. Genetic lesions of bilirubin uridine-diphosphoglucuronate glucuronosyltransferase(UGT1A1) causing Crigler-Najjar and Gilbert syndromes: correlation of genotype tophenotype. Hum Mutat. 2000;16(4):297–306. doi: 10.1002/1098-1004(200010)16:4<297::AID-HUMU2>3.0.CO;2-Z. - DOI - PubMed
-
- Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The human GeneMutation database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2014;133(1):1–9. doi: 10.1007/s00439-013-1358-4. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
