

Cellular adaptation to hypoxia through hypoxia inducible factors and beyond
- PMID: 32144406
- PMCID: PMC7222024
- DOI: 10.1038/s41580-020-0227-y
Cellular adaptation to hypoxia through hypoxia inducible factors and beyond
Abstract
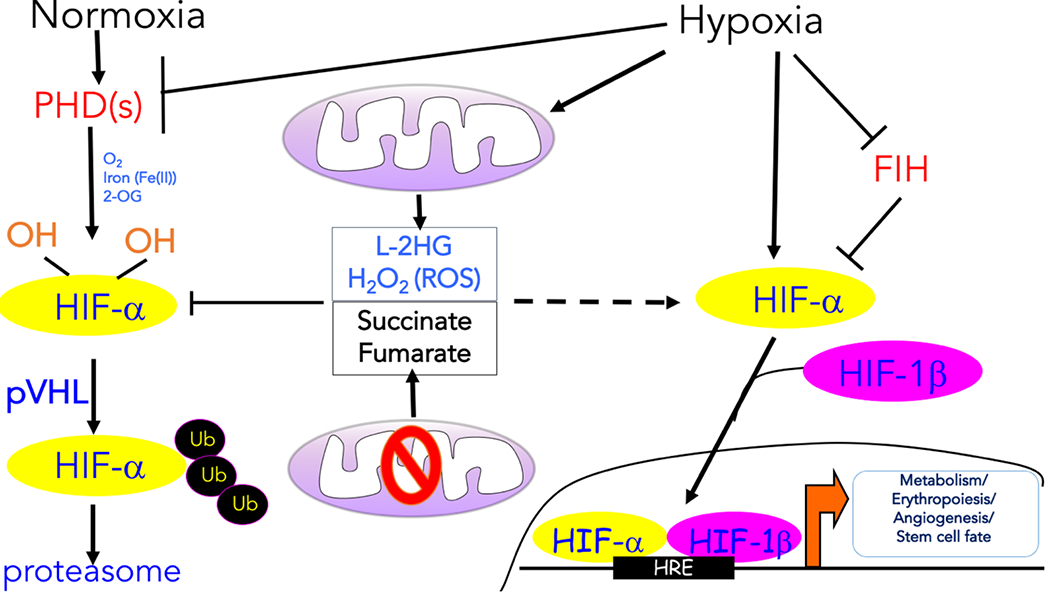
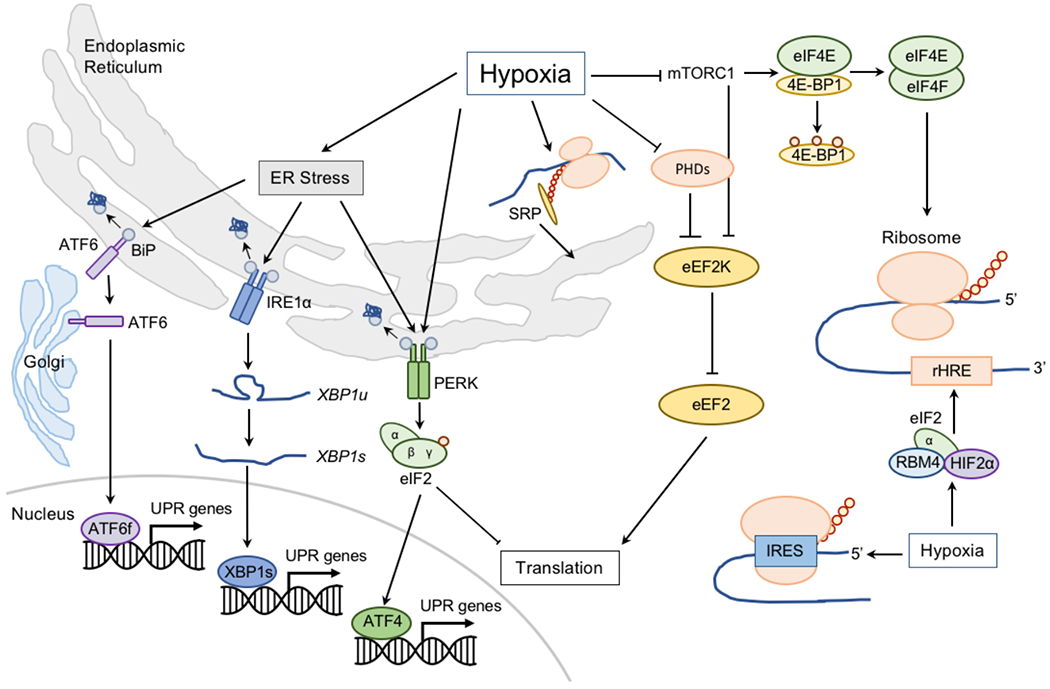
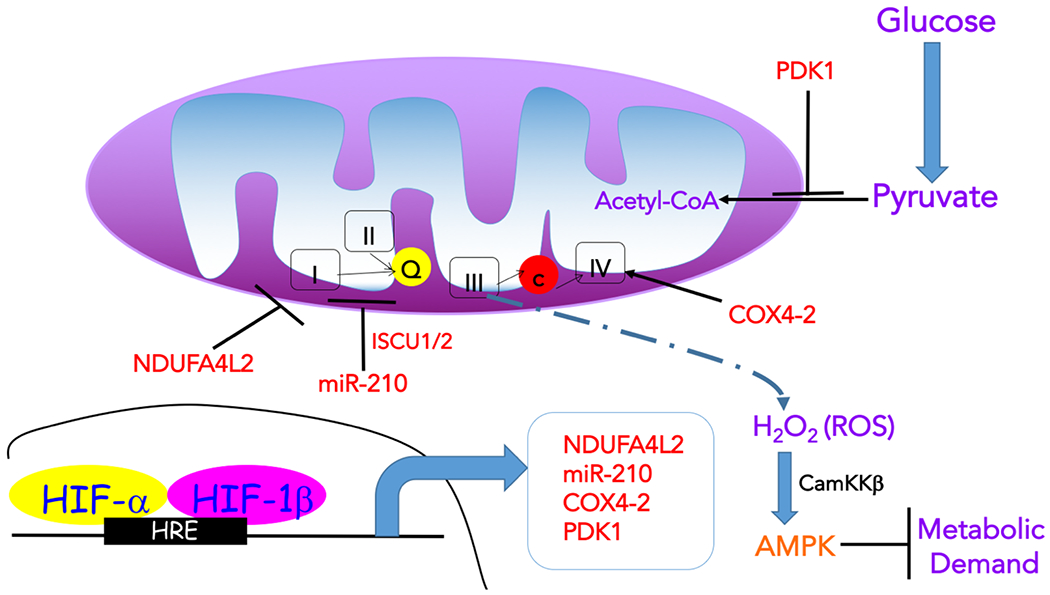
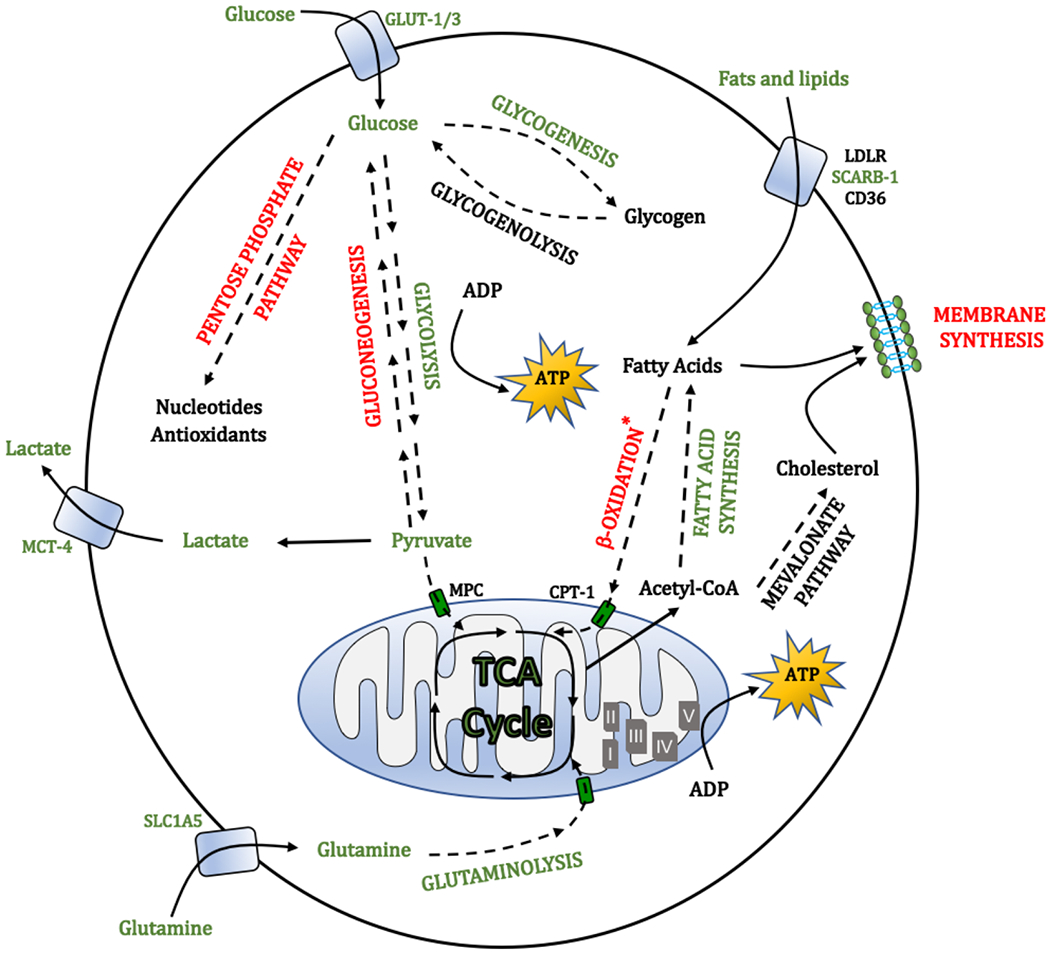
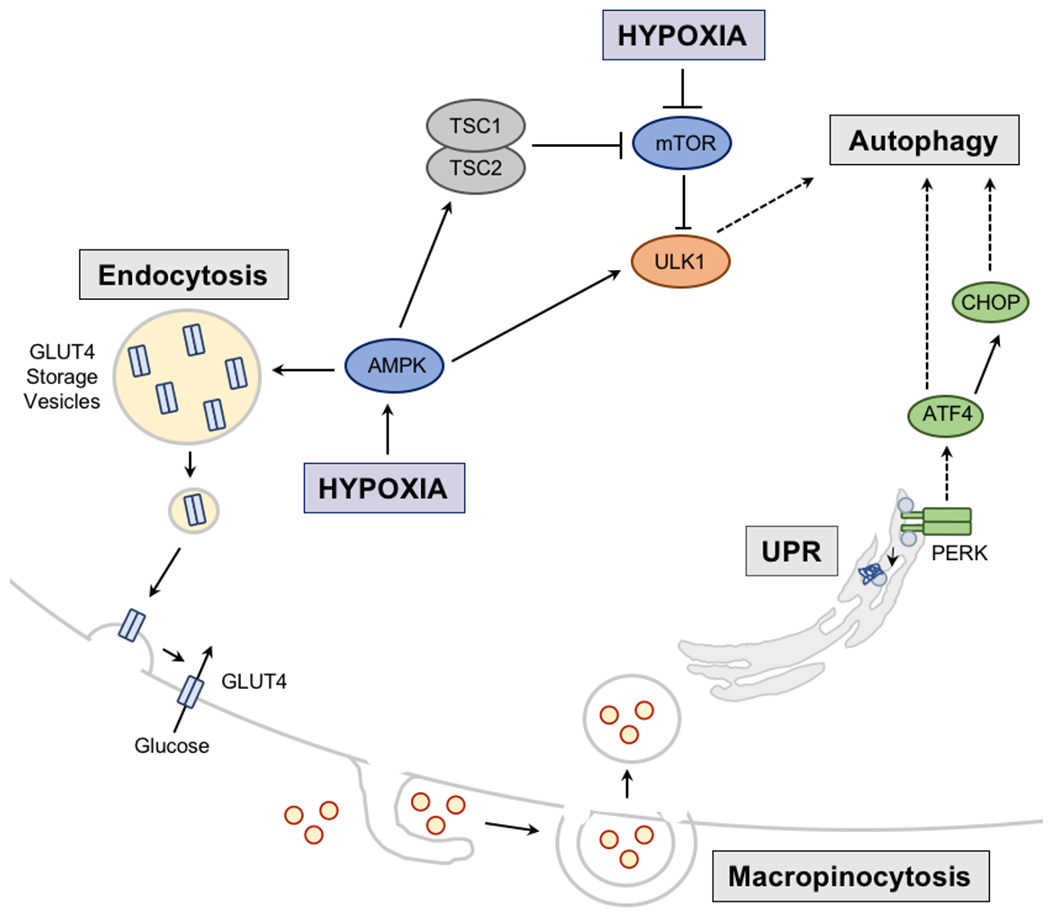
Molecular oxygen (O2) sustains intracellular bioenergetics and is consumed by numerous biochemical reactions, making it essential for most species on Earth. Accordingly, decreased oxygen concentration (hypoxia) is a major stressor that generally subverts life of aerobic species and is a prominent feature of pathological states encountered in bacterial infection, inflammation, wounds, cardiovascular defects and cancer. Therefore, key adaptive mechanisms to cope with hypoxia have evolved in mammals. Systemically, these adaptations include increased ventilation, cardiac output, blood vessel growth and circulating red blood cell numbers. On a cellular level, ATP-consuming reactions are suppressed, and metabolism is altered until oxygen homeostasis is restored. A critical question is how mammalian cells sense oxygen levels to coordinate diverse biological outputs during hypoxia. The best-studied mechanism of response to hypoxia involves hypoxia inducible factors (HIFs), which are stabilized by low oxygen availability and control the expression of a multitude of genes, including those involved in cell survival, angiogenesis, glycolysis and invasion/metastasis. Importantly, changes in oxygen can also be sensed via other stress pathways as well as changes in metabolite levels and the generation of reactive oxygen species by mitochondria. Collectively, this leads to cellular adaptations of protein synthesis, energy metabolism, mitochondrial respiration, lipid and carbon metabolism as well as nutrient acquisition. These mechanisms are integral inputs into fine-tuning the responses to hypoxic stress.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
References
-
- Pouyssegur J & López-Barneo J Hypoxia in health and disease. Molecular Aspects of Medicine 47–48, 1–2 (2016). - PubMed
-
- Kaelin WG et al. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 30, 393–402 (2008). - PubMed
-
Reviews by Semenza (2012) and Kaelin et al, (2008) provide an excellent outline of the regulation and importance of HIFs in physiology and diseases.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
