

Autonomic nervous system and inflammation interaction in endometriosis-associated pain
- PMID: 32145751
- PMCID: PMC7060607
- DOI: 10.1186/s12974-020-01752-1
Autonomic nervous system and inflammation interaction in endometriosis-associated pain
Abstract
Endometriosis is a chronic inflammatory disease. Pain is the most common symptom in endometriosis. Endometriosis-associated pain is caused by inflammation, and is related to aberrant innervation. Although the specific mechanism between endometriosis-associated pain and the interaction of aberrant innervation and inflammation remains unclear, many studies have confirmed certain correlations between them. In addition, we found that some chronic inflammatory autoimmune diseases (AIDs) such as inflammatory bowel disease (IBD) and rheumatoid arthritis (RA) share similar characteristics: the changes in dysregulation of inflammatory factors as well as the function and innervation of the autonomic nervous system (ANS). The mechanisms underlying the interaction between the ANS and inflammation have provided new advances among these disorders. Therefore, the purpose of this review is to compare the changes in inflammation and ANS in endometriosis, IBD, and RA; and to explore the role and possible mechanism of sympathetic and parasympathetic nerves in endometriosis-associated inflammation by referring to IBD and RA studies to provide some reference for further endometriosis research and treatment.
Keywords: Autonomic nervous system; Endometriosis; Inflammation; Pain.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures


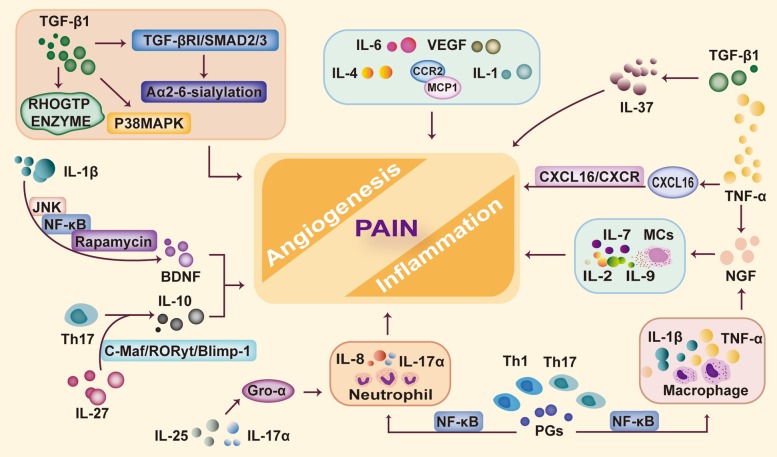
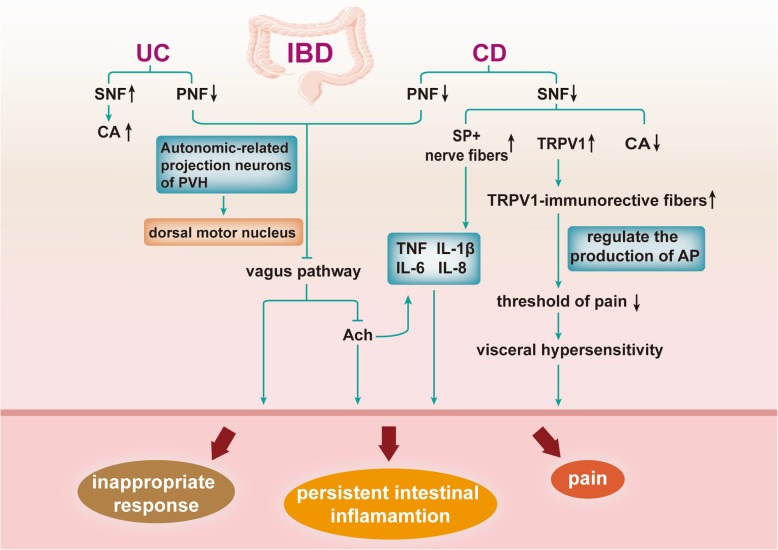
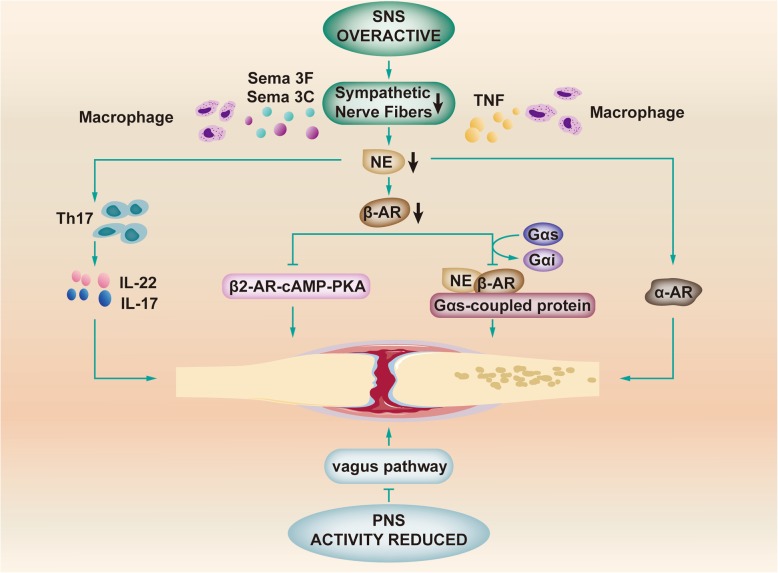
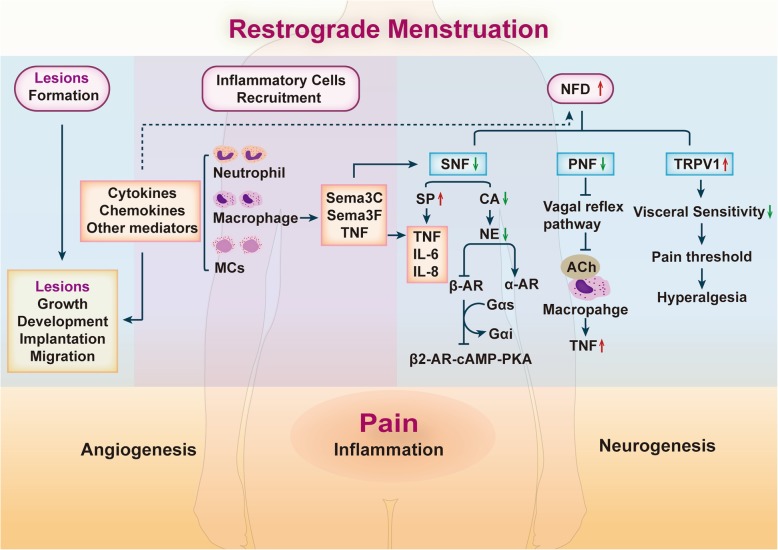
References
-
- Yu J, Francisco A, Patel BG, Cline JM, Zou E, Berga SL, Taylor RN. IL-1beta stimulates brain-derived neurotrophic factor production in eutopic endometriosis stromal cell cultures: a model for cytokine regulation of neuroangiogenesis. Am J Pathol. 2018;188:2281–2292. doi: 10.1016/j.ajpath.2018.06.011. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
- Grant No. 2016A030310151/Natural Science Foundation of Guangdong Province (CN)
- Grant No. 201901097/Student Innovation Training Program of Sun Yat-Sen University
- pdjh2019a0004/Special Funds for the Cultivation of Guangdong College Students' Scientific and Technological Innovation
- Grant No. 81701416/National Natural Science Foundation of China
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
