

Macrophages in obesity and non-alcoholic fatty liver disease: Crosstalk with metabolism
- PMID: 32149275
- PMCID: PMC7052781
- DOI: 10.1016/j.jhepr.2019.02.004
Macrophages in obesity and non-alcoholic fatty liver disease: Crosstalk with metabolism
Abstract
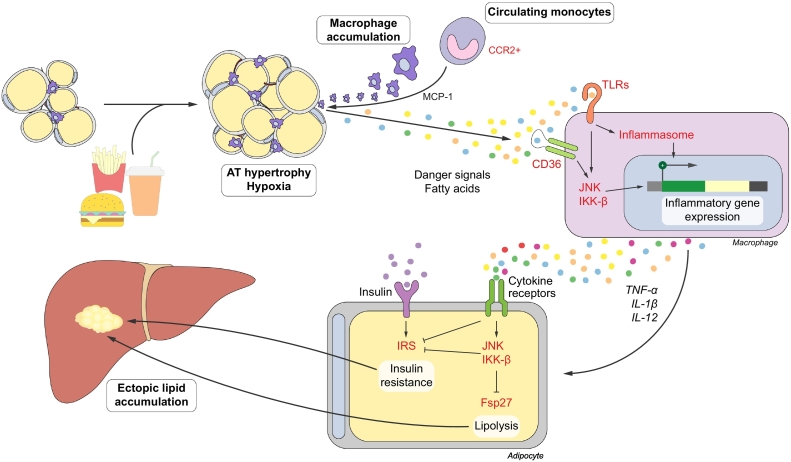
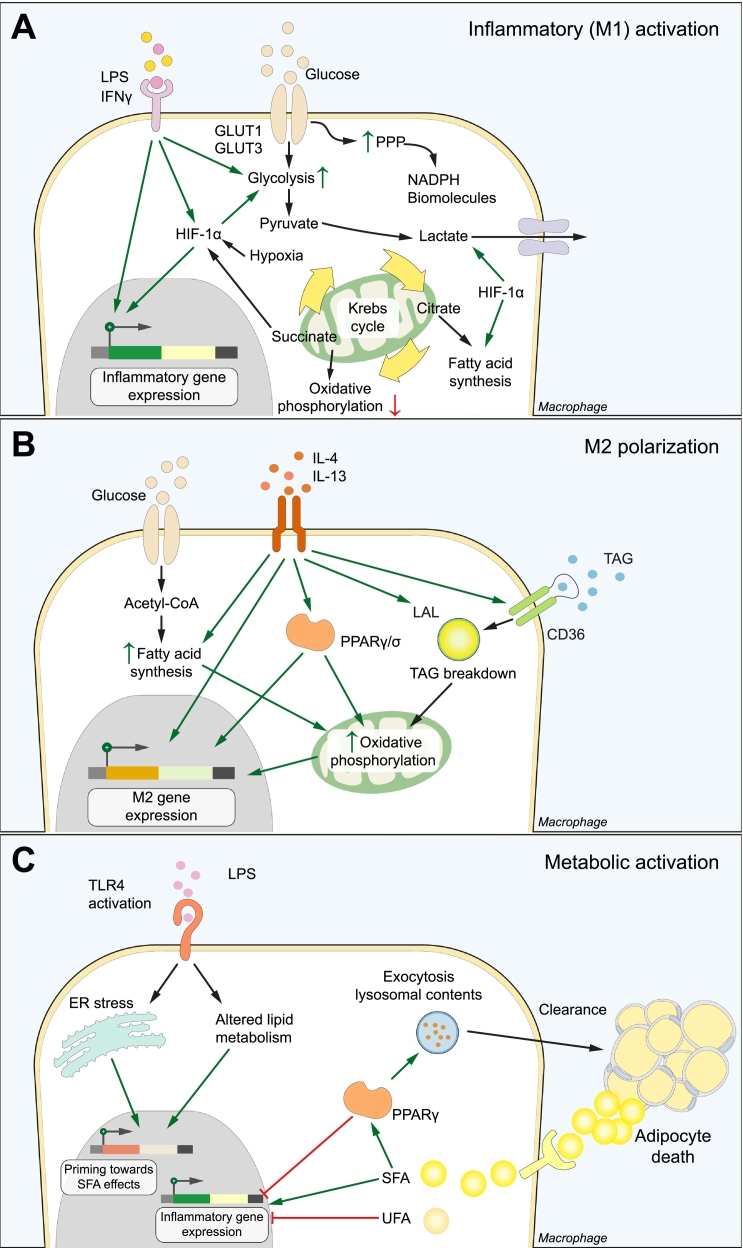
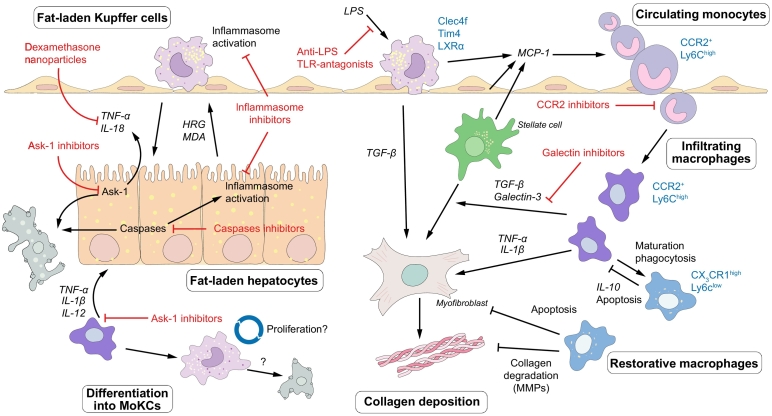
Non-alcoholic fatty liver disease (NAFLD) is the most prevalent liver disease worldwide, and a major cause of liver cirrhosis and hepatocellular carcinoma. NAFLD is intimately linked with other metabolic disorders characterized by insulin resistance. Metabolic diseases are driven by chronic inflammatory processes, in which macrophages perform essential roles. The polarization status of macrophages is itself influenced by metabolic stimuli such as fatty acids, which in turn affect the progression of metabolic dysfunction at multiple disease stages and in various tissues. For instance, adipose tissue macrophages respond to obesity, adipocyte stress and dietary factors by a specific metabolic and inflammatory programme that stimulates disease progression locally and in the liver. Kupffer cells and monocyte-derived macrophages represent ontologically distinct hepatic macrophage populations that perform a range of metabolic functions. These macrophages integrate signals from the gut-liver axis (related to dysbiosis, reduced intestinal barrier integrity, endotoxemia), from overnutrition, from systemic low-grade inflammation and from the local environment of a steatotic liver. This makes them central players in the progression of NAFLD to steatohepatitis (non-alcoholic steatohepatitis or NASH) and fibrosis. Moreover, the particular involvement of Kupffer cells in lipid metabolism, as well as the inflammatory activation of hepatic macrophages, may pathogenically link NAFLD/NASH and cardiovascular disease. In this review, we highlight the polarization, classification and function of macrophage subsets and their interaction with metabolic cues in the pathophysiology of obesity and NAFLD. Evidence from animal and clinical studies suggests that macrophage targeting may improve the course of NAFLD and related metabolic disorders.
Keywords: NASH; immunometabolism; innate immunity; liver immunology; steatohepatitis, Kupffer cells.
© 2019 The Author(s).
Figures
References
-
- Yki-Jarvinen H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014;2:901–910. - PubMed
-
- Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15:11–20. - PubMed
-
- Ekstedt M, Hagstrom H, Nasr P, Fredrikson M, Stal P, Kechagias S. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology. 2015;61:1547–1554. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
