

A putative silencer variant in a spontaneous canine model of retinitis pigmentosa
- PMID: 32150541
- PMCID: PMC7082071
- DOI: 10.1371/journal.pgen.1008659
A putative silencer variant in a spontaneous canine model of retinitis pigmentosa
Abstract
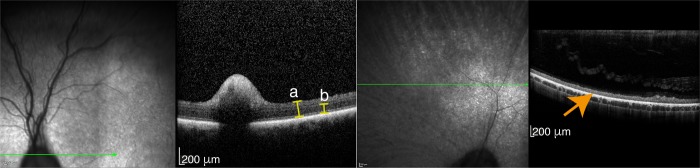
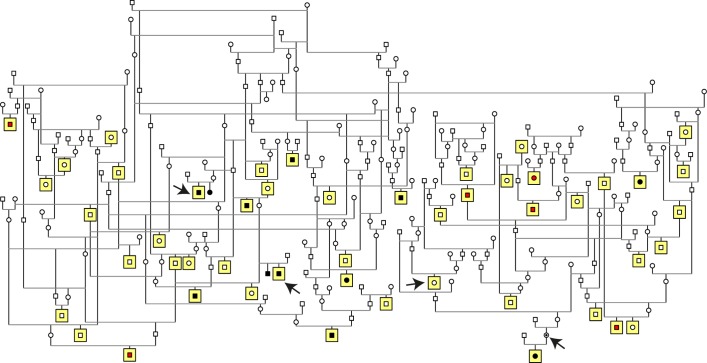
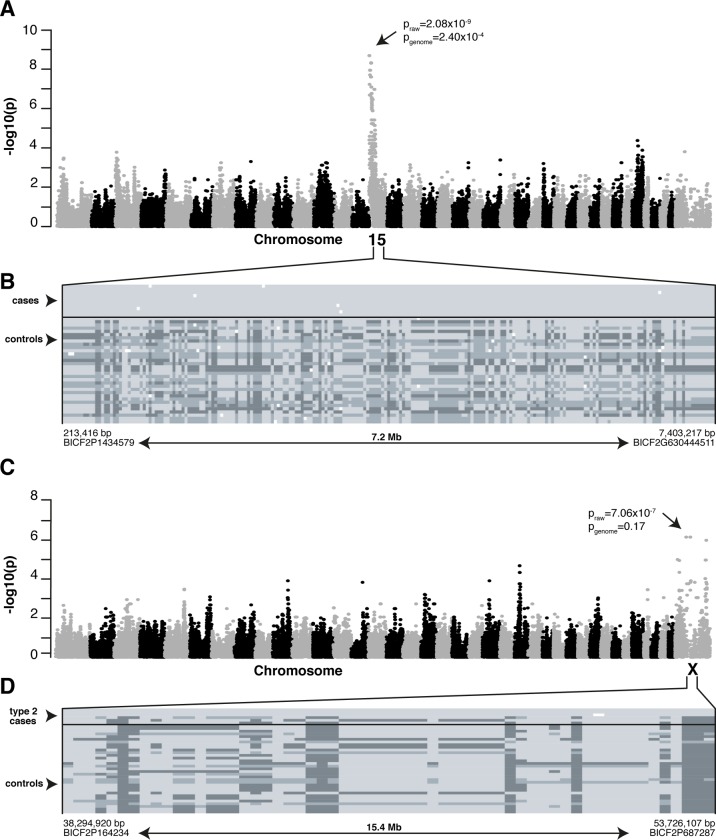
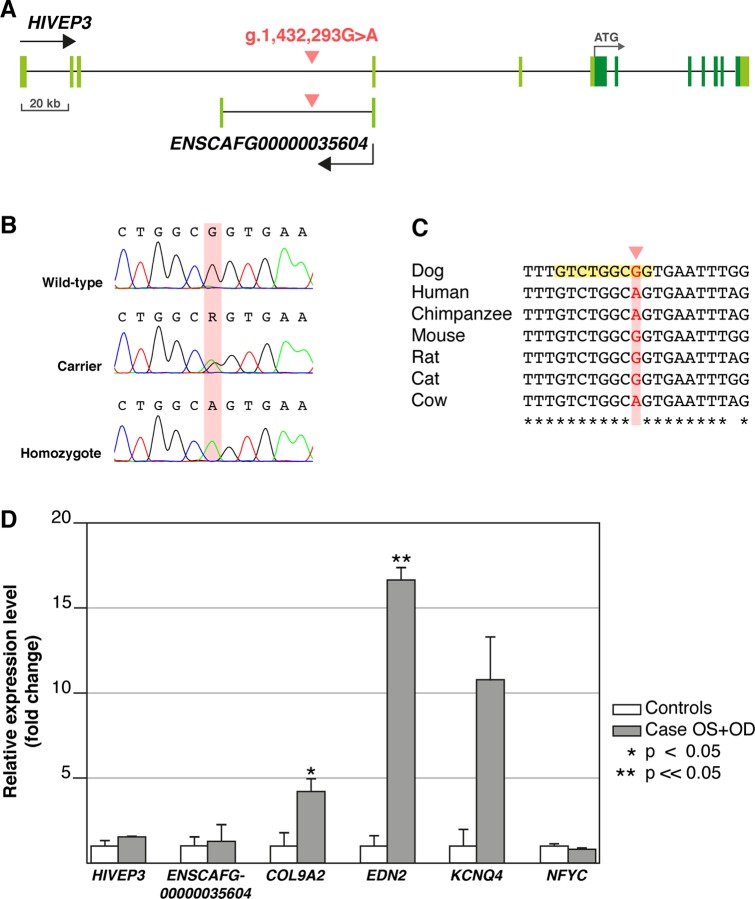
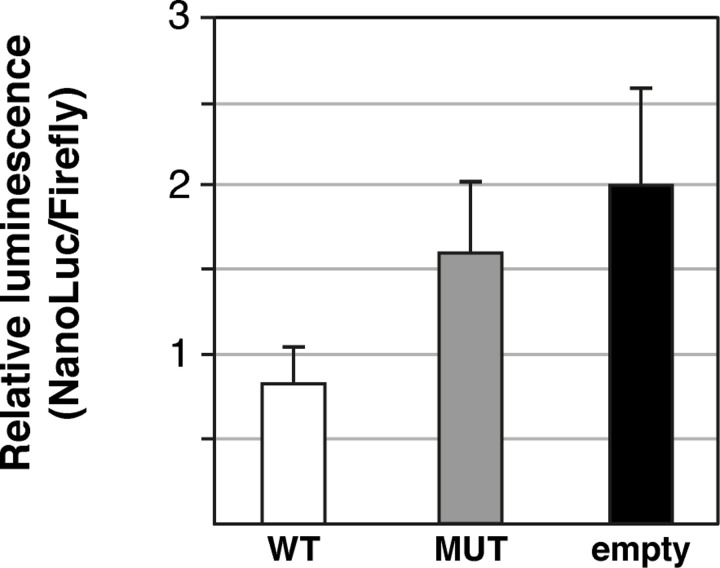
Retinitis pigmentosa (RP) is the leading cause of blindness with nearly two million people affected worldwide. Many genes have been implicated in RP, yet in 30-80% of the RP patients the genetic cause remains unknown. A similar phenotype, progressive retinal atrophy (PRA), affects many dog breeds including the Miniature Schnauzer. We performed clinical, genetic and functional experiments to identify the genetic cause of PRA in the breed. The age of onset and pattern of disease progression suggested that at least two forms of PRA, types 1 and 2 respectively, affect the breed, which was confirmed by genome-wide association study that implicated two distinct genomic loci in chromosomes 15 and X, respectively. Whole-genome sequencing revealed a fully segregating recessive regulatory variant in type 1 PRA. The associated variant has a very recent origin based on haplotype analysis and lies within a regulatory site with the predicted binding site of HAND1::TCF3 transcription factor complex. Luciferase assays suggested that mutated regulatory sequence increases expression. Case-control retinal expression comparison of six best HAND1::TCF3 target genes were analyzed with quantitative reverse-transcriptase PCR assay and indicated overexpression of EDN2 and COL9A2 in the affected retina. Defects in both EDN2 and COL9A2 have been previously associated with retinal degeneration. In summary, our study describes two genetically different forms of PRA and identifies a fully penetrant variant in type 1 form with a possible regulatory effect. This would be among the first reports of a regulatory variant in retinal degeneration in any species, and establishes a new spontaneous dog model to improve our understanding of retinal biology and gene regulation while the affected breed will benefit from a reliable genetic testing.
Conflict of interest statement
We have read the journal’s policy and the authors of this manuscript have the following competing interests: HL is a paid consultant to Genoscoper Laboratories Ltd, which will provide a genetic test for the type 1 PRA in MSs.
Figures
Comment in
-
Formal commentary.PLoS Genet. 2020 Nov 5;16(11):e1009059. doi: 10.1371/journal.pgen.1009059. eCollection 2020 Nov. PLoS Genet. 2020. PMID: 33151924 Free PMC article. No abstract available.
References
-
- Daiger S, Rossiter B, Greenberg J, Christoffels A, Hide W. Data services and software for identifying genes and mutations causing retinal degeneration. Invest Ophthalmol Vis Sci 1998;39:S295.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
