

The application of RNA sequencing for the diagnosis and genomic classification of pediatric acute lymphoblastic leukemia
- PMID: 32150610
- PMCID: PMC7065479
- DOI: 10.1182/bloodadvances.2019001008
The application of RNA sequencing for the diagnosis and genomic classification of pediatric acute lymphoblastic leukemia
Erratum in
-
Brown LM, Lonsdale A, Zhu A, et al. The application of RNA sequencing for the diagnosis and genomic classification of pediatric acute lymphoblastic leukemia. Blood Adv. 2020;4(5):930-942.Blood Adv. 2020 Apr 14;4(7):1217. doi: 10.1182/bloodadvances.2020001859. Blood Adv. 2020. PMID: 32227206 Free PMC article. No abstract available.
Abstract
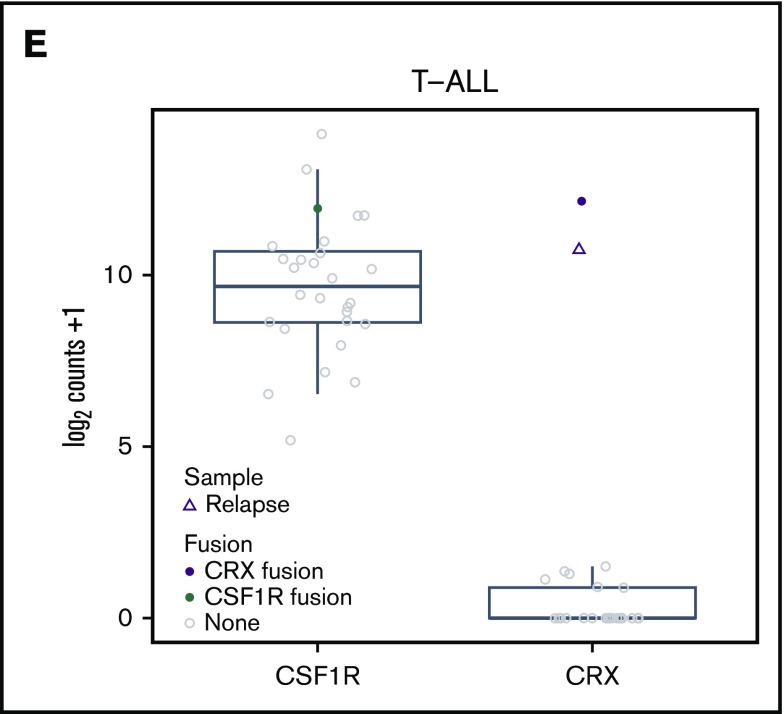
Acute lymphoblastic leukemia (ALL) is the most common childhood malignancy, and implementation of risk-adapted therapy has been instrumental in the dramatic improvements in clinical outcomes. A key to risk-adapted therapies includes the identification of genomic features of individual tumors, including chromosome number (for hyper- and hypodiploidy) and gene fusions, notably ETV6-RUNX1, TCF3-PBX1, and BCR-ABL1 in B-cell ALL (B-ALL). RNA-sequencing (RNA-seq) of large ALL cohorts has expanded the number of recurrent gene fusions recognized as drivers in ALL, and identification of these new entities will contribute to refining ALL risk stratification. We used RNA-seq on 126 ALL patients from our clinical service to test the utility of including RNA-seq in standard-of-care diagnostic pipelines to detect gene rearrangements and IKZF1 deletions. RNA-seq identified 86% of rearrangements detected by standard-of-care diagnostics. KMT2A (MLL) rearrangements, although usually identified, were the most commonly missed by RNA-seq as a result of low expression. RNA-seq identified rearrangements that were not detected by standard-of-care testing in 9 patients. These were found in patients who were not classifiable using standard molecular assessment. We developed an approach to detect the most common IKZF1 deletion from RNA-seq data and validated this using an RQ-PCR assay. We applied an expression classifier to identify Philadelphia chromosome-like B-ALL patients. T-ALL proved a rich source of novel gene fusions, which have clinical implications or provide insights into disease biology. Our experience shows that RNA-seq can be implemented within an individual clinical service to enhance the current molecular diagnostic risk classification of ALL.
© 2020 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures






References
-
- Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin. 2014;64(2):83-103. - PubMed
-
- Roberts KG, Mullighan CG. Genomics in acute lymphoblastic leukaemia: insights and treatment implications. Nat Rev Clin Oncol. 2015;12(6):344-357. - PubMed
-
- Mullighan CG, Goorha S, Radtke I, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446(7137):758-764. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
