

Identifying and Classifying Shared Selective Sweeps from Multilocus Data
- PMID: 32152048
- PMCID: PMC7198270
- DOI: 10.1534/genetics.120.303137
Identifying and Classifying Shared Selective Sweeps from Multilocus Data
Abstract
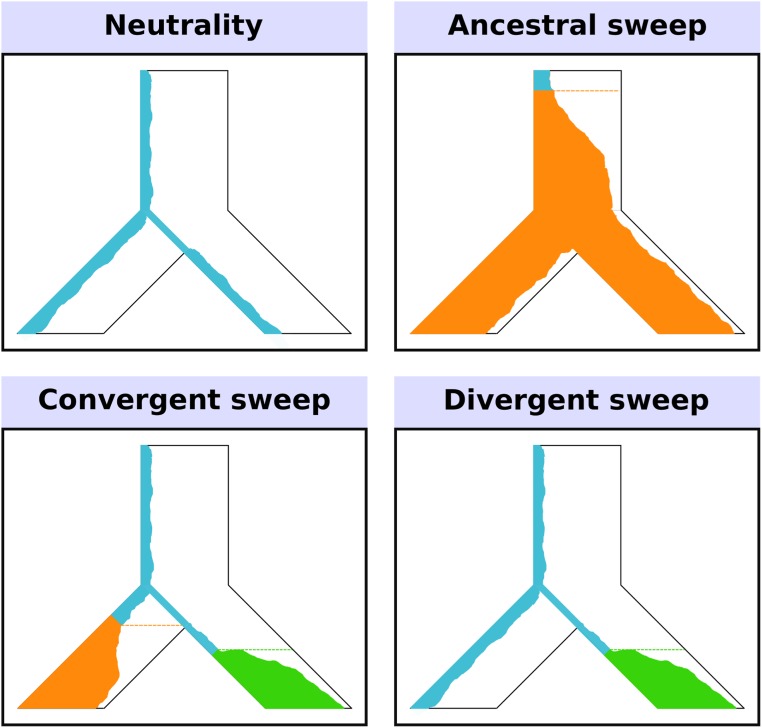
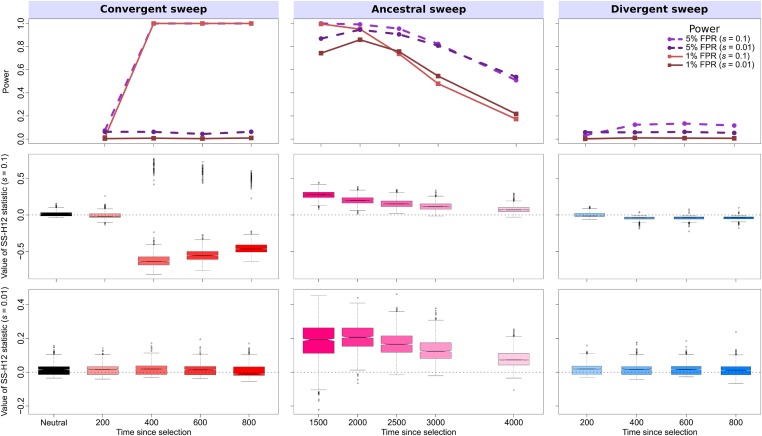
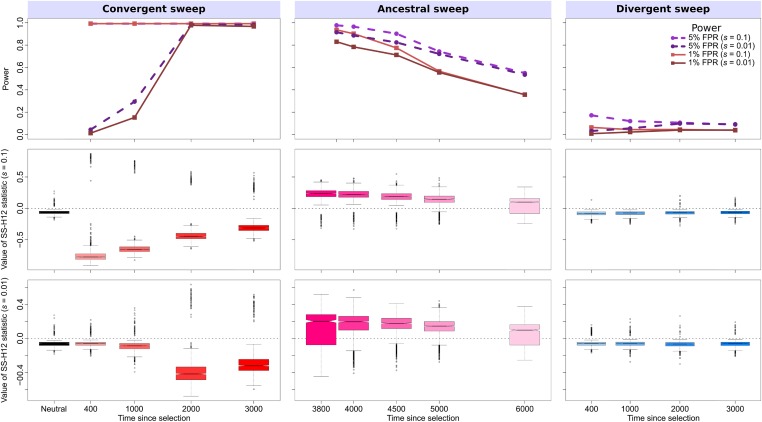
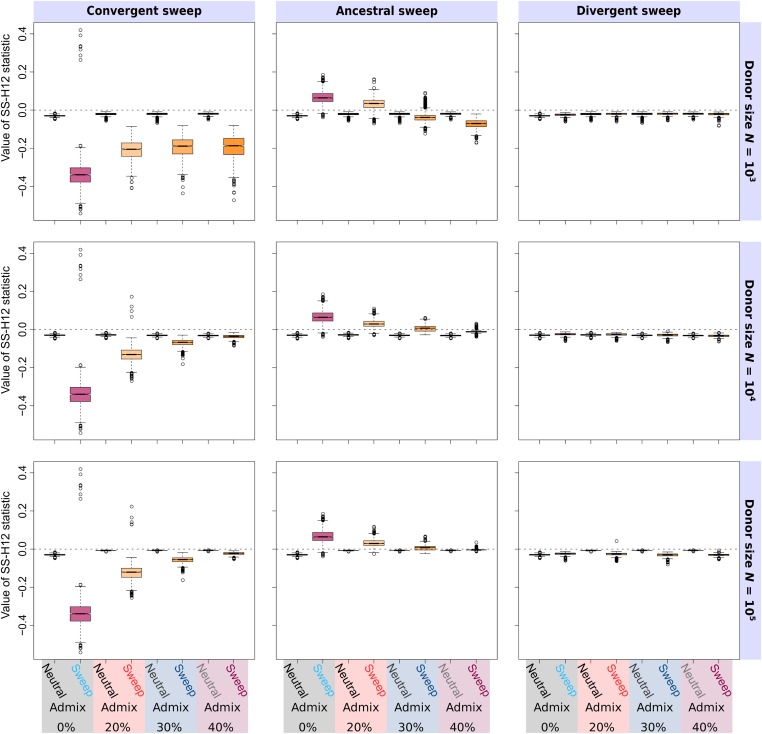
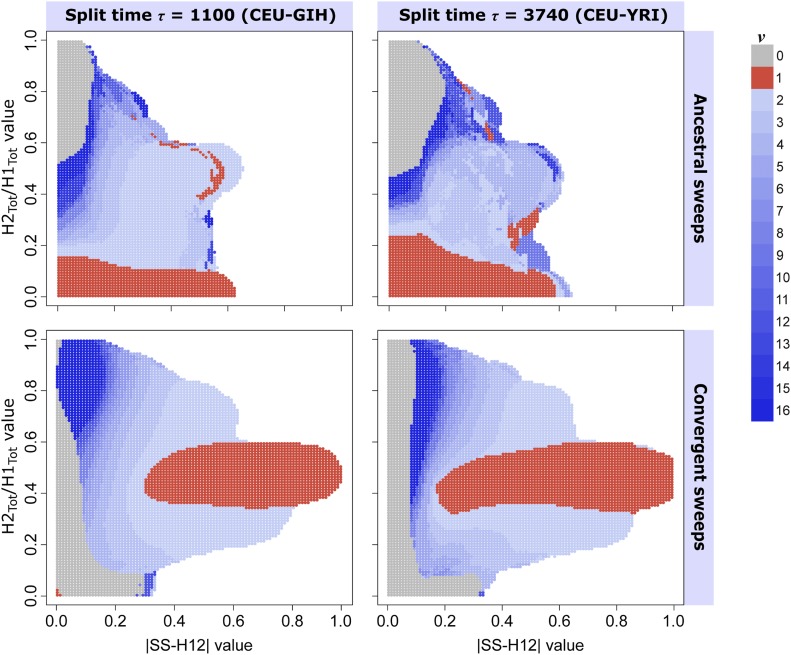
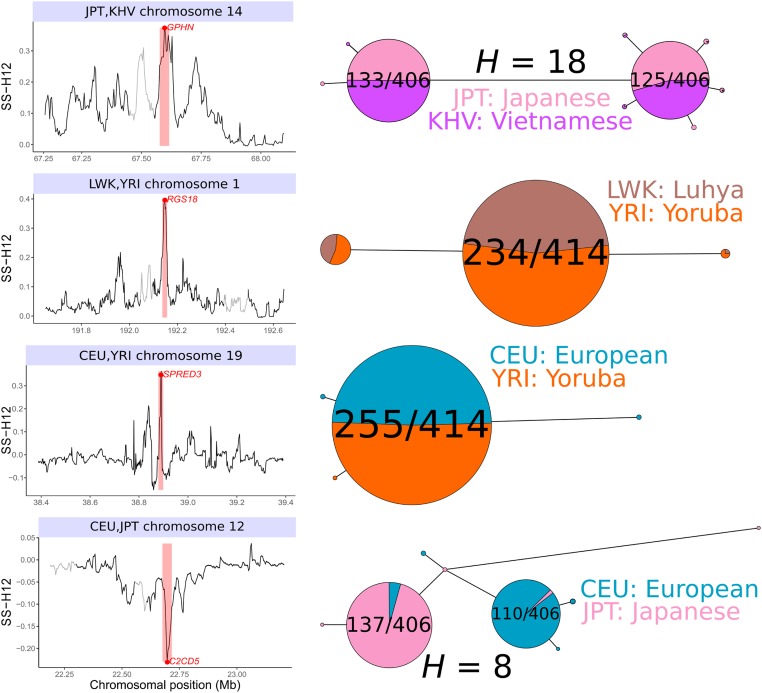
Positive selection causes beneficial alleles to rise to high frequency, resulting in a selective sweep of the diversity surrounding the selected sites. Accordingly, the signature of a selective sweep in an ancestral population may still remain in its descendants. Identifying signatures of selection in the ancestor that are shared among its descendants is important to contextualize the timing of a sweep, but few methods exist for this purpose. We introduce the statistic SS-H12, which can identify genomic regions under shared positive selection across populations and is based on the theory of the expected haplotype homozygosity statistic H12, which detects recent hard and soft sweeps from the presence of high-frequency haplotypes. SS-H12 is distinct from comparable statistics because it requires a minimum of only two populations, and properly identifies and differentiates between independent convergent sweeps and true ancestral sweeps, with high power and robustness to a variety of demographic models. Furthermore, we can apply SS-H12 in conjunction with the ratio of statistics we term [Formula: see text] and [Formula: see text] to further classify identified shared sweeps as hard or soft. Finally, we identified both previously reported and novel shared sweep candidates from human whole-genome sequences. Previously reported candidates include the well-characterized ancestral sweeps at LCT and SLC24A5 in Indo-Europeans, as well as GPHN worldwide. Novel candidates include an ancestral sweep at RGS18 in sub-Saharan Africans involved in regulating the platelet response and implicated in sudden cardiac death, and a convergent sweep at C2CD5 between European and East Asian populations that may explain their different insulin responses.
Keywords: ancestral sweep; convergent sweep; expected haplotype homozygosity; multilocus genotype.
Copyright © 2020 by the Genetics Society of America.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
