

Programmed axon degeneration: from mouse to mechanism to medicine
- PMID: 32152523
- PMCID: PMC8926152
- DOI: 10.1038/s41583-020-0269-3
Programmed axon degeneration: from mouse to mechanism to medicine
Abstract
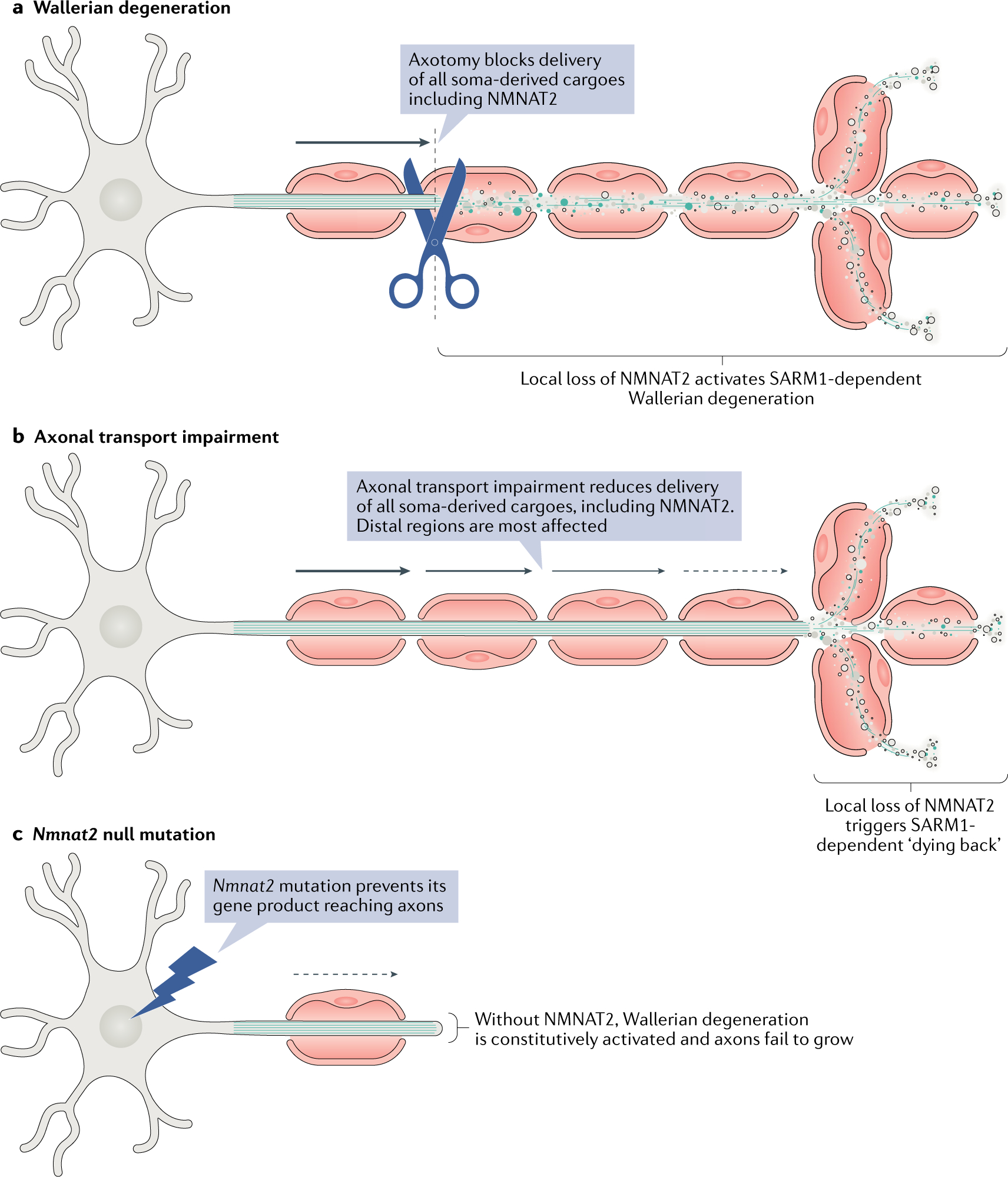
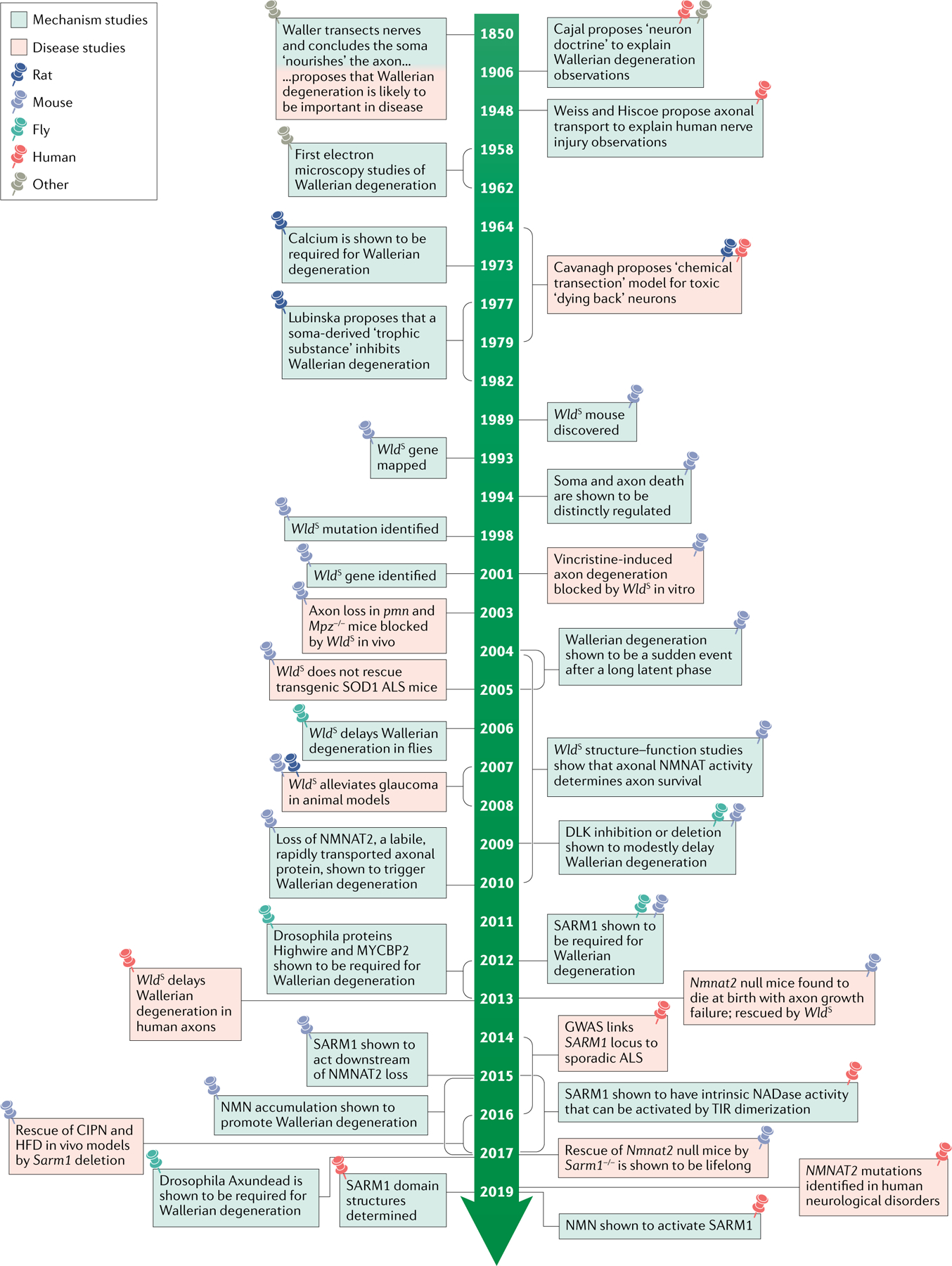
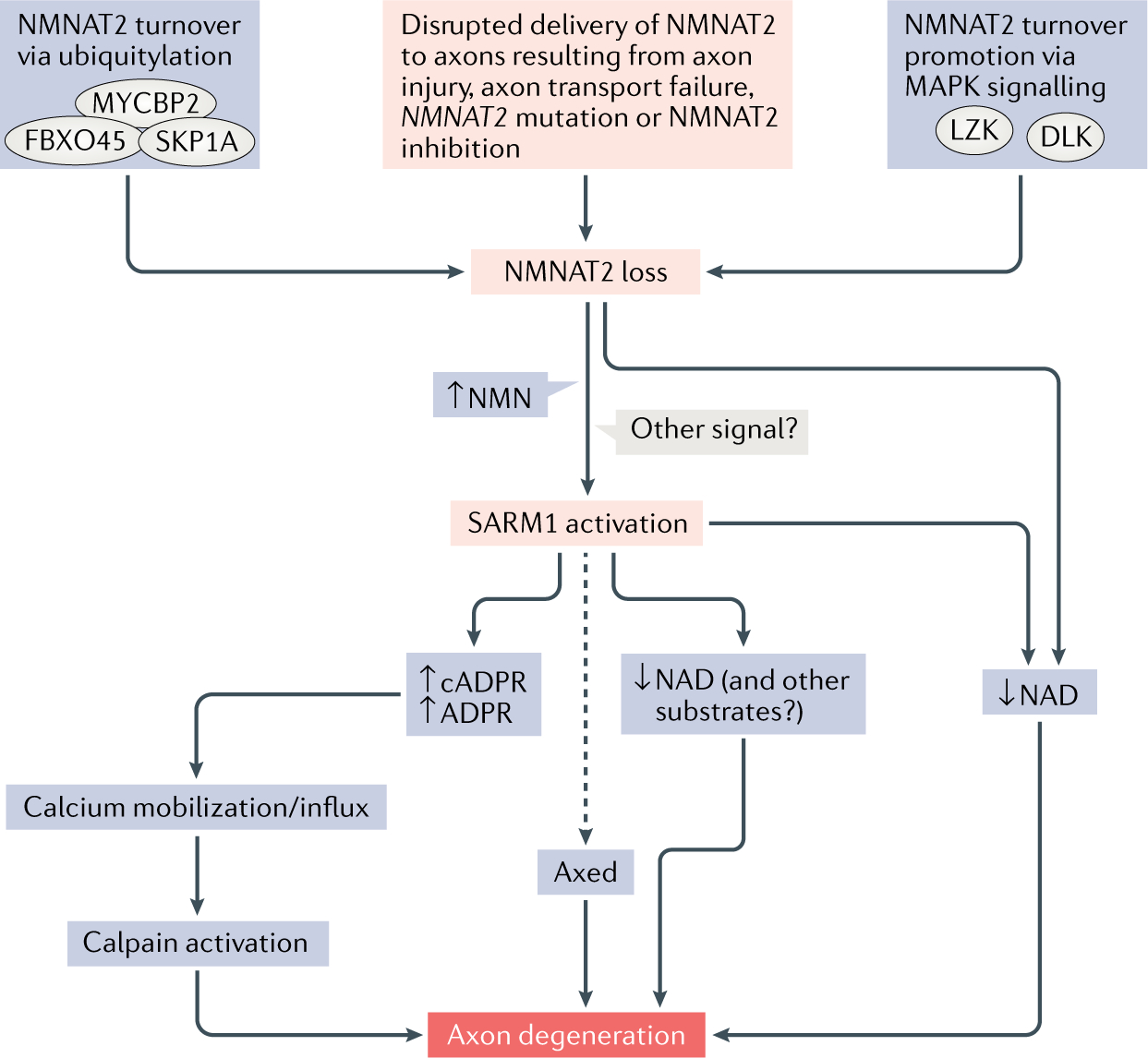
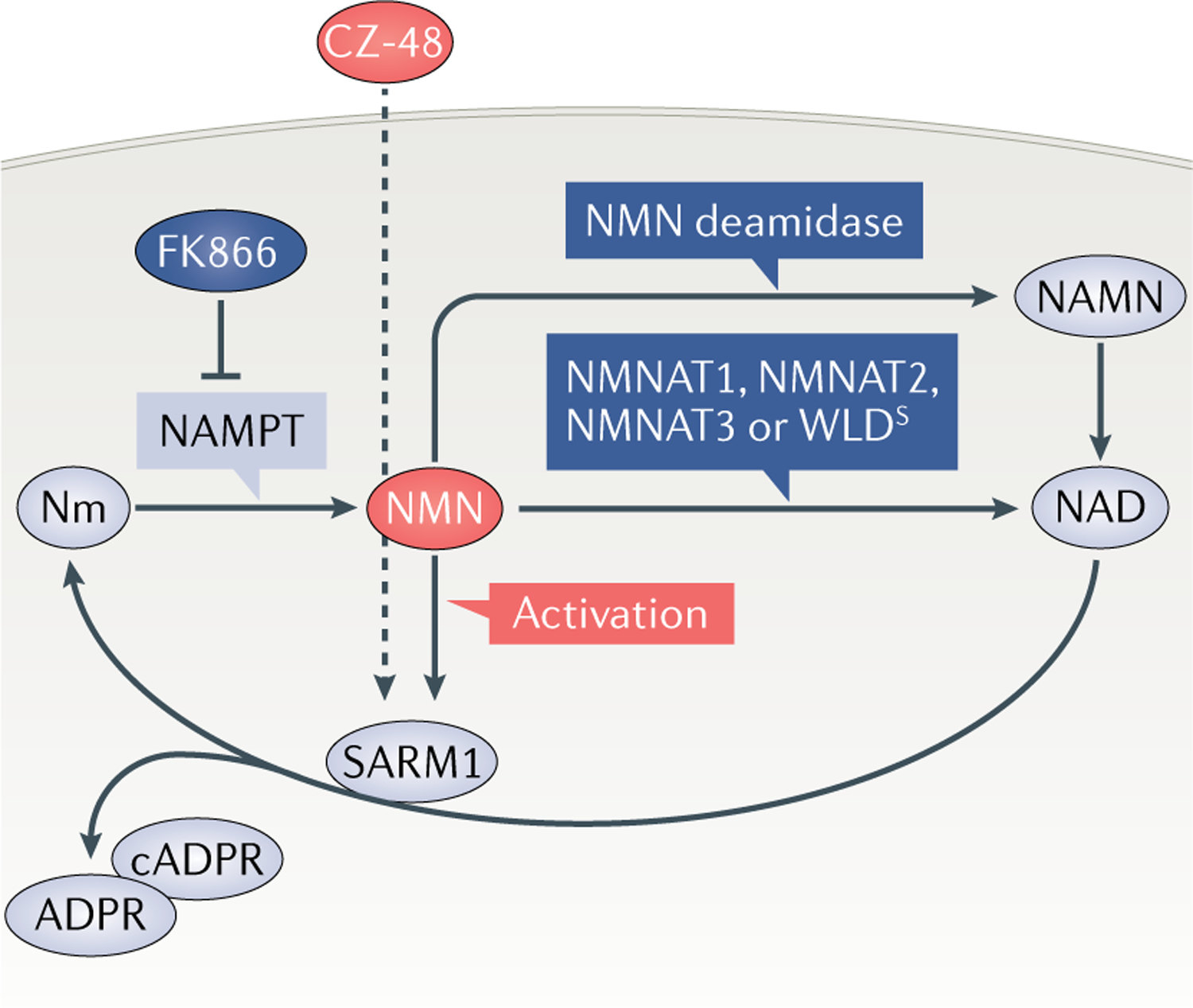
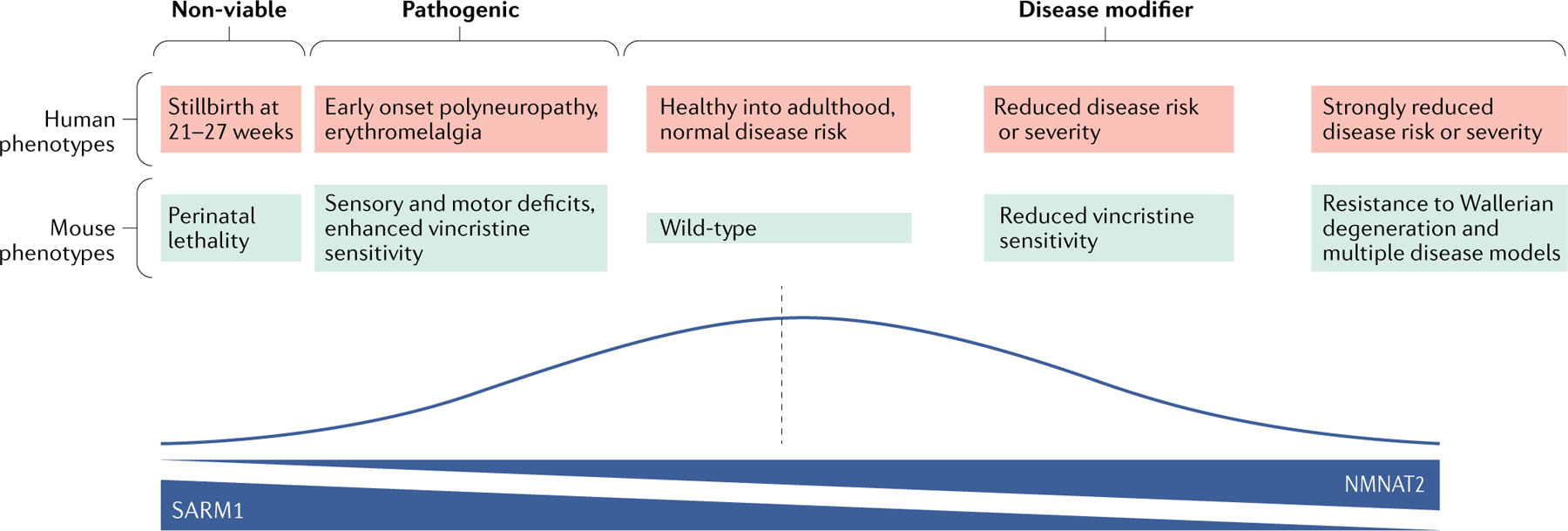
Wallerian degeneration is a widespread mechanism of programmed axon degeneration. In the three decades since the discovery of the Wallerian degeneration slow (WldS) mouse, research has generated extensive knowledge of the molecular mechanisms underlying Wallerian degeneration, demonstrated its involvement in non-injury disorders and found multiple ways to block it. Recent developments have included: the detection of NMNAT2 mutations that implicate Wallerian degeneration in rare human diseases; the capacity for lifelong rescue of a lethal condition related to Wallerian degeneration in mice; the discovery of 'druggable' enzymes, including SARM1 and MYCBP2 (also known as PHR1), in Wallerian pathways; and the elucidation of protein structures to drive further understanding of the underlying mechanisms and drug development. Additionally, new data have indicated the potential of these advances to alleviate a number of common disorders, including chemotherapy-induced and diabetic peripheral neuropathies, traumatic brain injury, and amyotrophic lateral sclerosis.
Conflict of interest statement
Competing interests
M.P.C. has an academic collaboration with AstraZeneca and is a consultant for Proneurotech. A.H. serves on the scientific advisory board of Disarm Therapeutics.
Figures
References
-
- Waller A Experiments on the section of the glossopharyngeal and hypoglossal nerves of the frog, and observations of the alterations produced thereby in the structure of their primitive fibres. Philos. Trans. R. Soc. Lond. 140, 423–429 (1850).
-
-
Gilley J, Ribchester RR & Coleman M R Sarm 1 deletion, but not Wlds, confers lifelong rescue in a mouse model of severe axonopathy. Cell Rep. 21, 10–16(2017).
This study shows that blocking Wallerian degeneration can permanently rescue axons in some circumstances.
-
-
- Conforti L, Gilley J & Coleman M R Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat. Rev. Neurosci. 15, 394–409 (2014). - PubMed
-
-
Lunn ER, Perry VH, Brown MC, Rosen H & Gordon S Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. Eur. J. Neurosci. 1,27–33 (1989).
The discovery of the Wlds (formerly ‘Ola’) mouse, which initiated a molecular understanding of Wallerian degeneration.
-
-
- Lubińska L Patterns of Wallerian degeneration of myelinated fibres in short and long peripheral stumps and in isolated segments of rat phrenic nerve. Interpretation of the role of axoplasmic flow of the trophic factor. Brain Res. 233, 227–240 (1982). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
