

CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine
- PMID: 32153386
- PMCID: PMC7046560
- DOI: 10.3389/fphar.2019.01662
CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine
Abstract
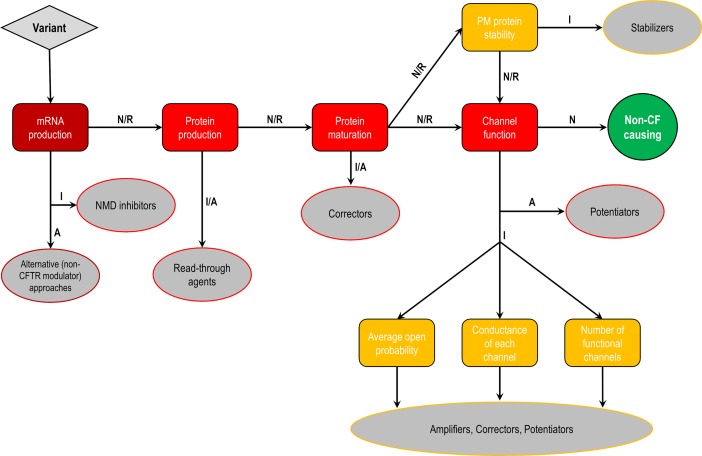
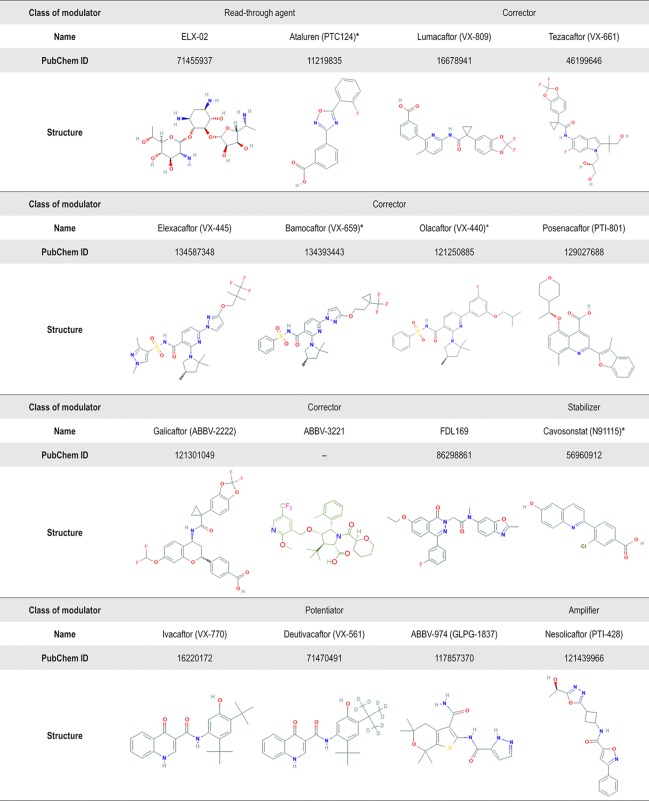
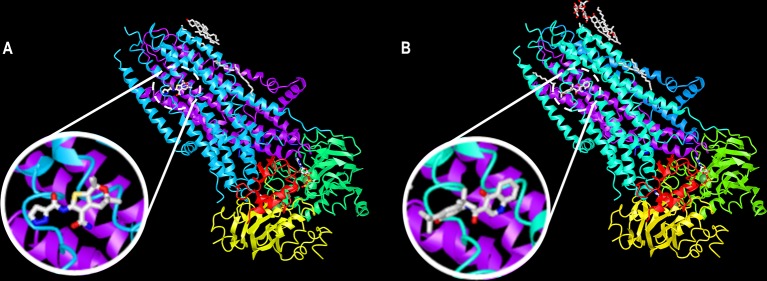
Cystic fibrosis (CF) is a lethal inherited disease caused by mutations in the CF transmembrane conductance regulator (CFTR) gene, which result in impairment of CFTR mRNA and protein expression, function, stability or a combination of these. Although CF leads to multifaceted clinical manifestations, the respiratory disorder represents the major cause of morbidity and mortality of these patients. The life expectancy of CF patients has substantially lengthened due to early diagnosis and improvements in symptomatic therapeutic regimens. Quality of life remains nevertheless limited, as these individuals are subjected to considerable clinical, psychosocial and economic burdens. Since the discovery of the CFTR gene in 1989, tremendous efforts have been made to develop therapies acting more upstream on the pathogenesis cascade, thereby overcoming the underlying dysfunctions caused by CFTR mutations. In this line, the advances in cell-based high-throughput screenings have been facilitating the fast-tracking of CFTR modulators. These modulator drugs have the ability to enhance or even restore the functional expression of specific CF-causing mutations, and they have been classified into five main groups depending on their effects on CFTR mutations: potentiators, correctors, stabilizers, read-through agents, and amplifiers. To date, four CFTR modulators have reached the market, and these pharmaceutical therapies are transforming patients' lives with short- and long-term improvements in clinical outcomes. Such breakthroughs have paved the way for the development of novel CFTR modulators, which are currently under experimental and clinical investigations. Furthermore, recent insights into the CFTR structure will be useful for the rational design of next-generation modulator drugs. This review aims to provide a summary of recent developments in CFTR-directed therapeutics. Barriers and future directions are also discussed in order to optimize treatment adherence, identify feasible and sustainable solutions for equitable access to these therapies, and continue to expand the pipeline of novel modulators that may result in effective precision medicine for all individuals with CF.
Keywords: CFTR mutations; cell models; clinical trials; cystic fibrosis; drug development; high-throughput screening; lung; personalized medicine.
Copyright © 2020 Lopes-Pacheco.
Figures


References
-
- Ahner A., Gong X., Schmidt B. Z., Peters K. W., Rabeh W. M., Thibodeau P. H., et al. (2013). Small heat shock proteins target cystic fibrosis transmembrane conductance regulator for degradation via a small ubiquitin-like modifier-dependent pathway. Mol. Biol. Cell. 24 (2), 74–84. 10.1091/mbc.e12-09-0678 - DOI - PMC - PubMed
-
- Alshafie W., Chappe F. G., Li M., Anini Y., Chappe V. M. (2014). VIP regulates CFTR membrane expression and function in Calu-3 cells by increasing its interaction with NHERF1 and P-ERM in a VPAC1- and PKCϵ-dependent manner. Am. J. Physiol. Cell Physiol. 307 (1), C107–C119. 10.1152/ajpcell.00296.2013 - DOI - PubMed
-
- Amaral M. D., de Boeck K., ECFS Strategic Planning Task Force on ‘Speeding up access to new drugs for CF' (2019). Theranostics by testing CFTR modulators in patients-derived materials: The current status and a proposal for subjects with rare CFTR mutations. J. Cyst. Fibros. 18 (5), 685–6925. 10.1016/j.jcf.2019.06.010 - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical