

A Guide to Carrying Out a Phylogenomic Target Sequence Capture Project
- PMID: 32153629
- PMCID: PMC7047930
- DOI: 10.3389/fgene.2019.01407
A Guide to Carrying Out a Phylogenomic Target Sequence Capture Project
Abstract
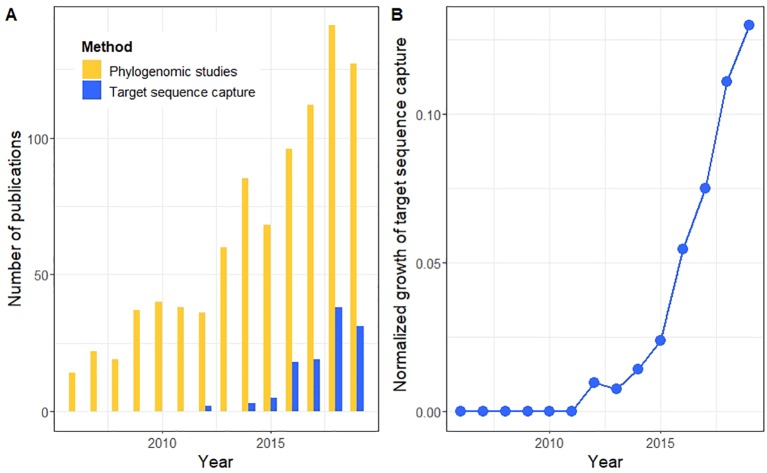
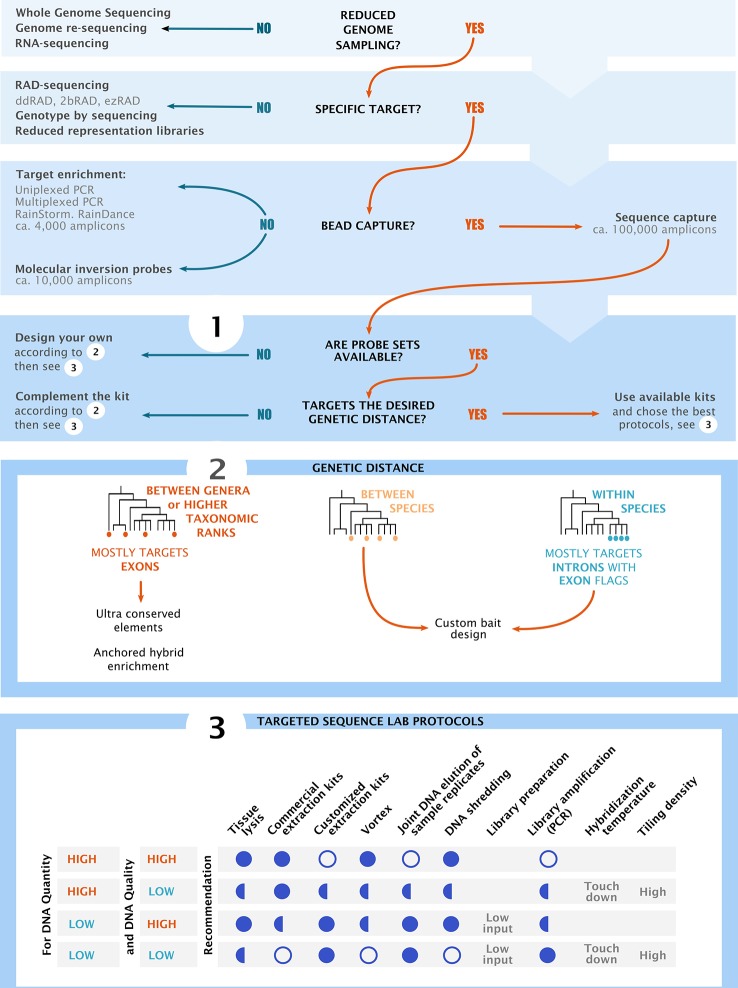
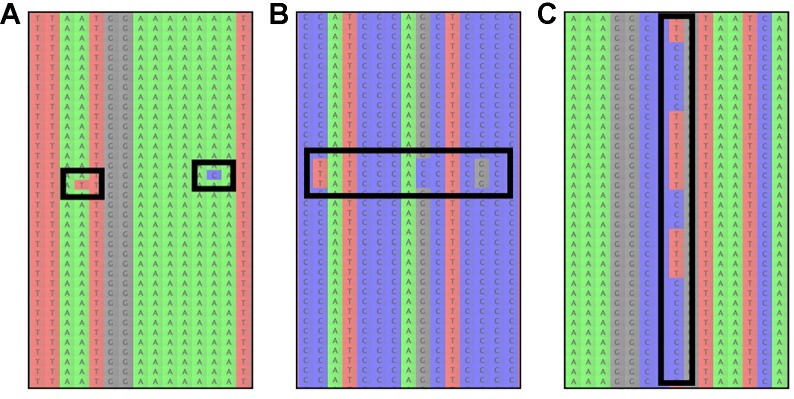
High-throughput DNA sequencing techniques enable time- and cost-effective sequencing of large portions of the genome. Instead of sequencing and annotating whole genomes, many phylogenetic studies focus sequencing effort on large sets of pre-selected loci, which further reduces costs and bioinformatic challenges while increasing coverage. One common approach that enriches loci before sequencing is often referred to as target sequence capture. This technique has been shown to be applicable to phylogenetic studies of greatly varying evolutionary depth. Moreover, it has proven to produce powerful, large multi-locus DNA sequence datasets suitable for phylogenetic analyses. However, target capture requires careful considerations, which may greatly affect the success of experiments. Here we provide a simple flowchart for designing phylogenomic target capture experiments. We discuss necessary decisions from the identification of target loci to the final bioinformatic processing of sequence data. We outline challenges and solutions related to the taxonomic scope, sample quality, and available genomic resources of target capture projects. We hope this review will serve as a useful roadmap for designing and carrying out successful phylogenetic target capture studies.
Keywords: Hyb-Seq; Illumina; NGS; anchored enrichment; bait; high throughput sequencing; molecular phylogenetics; probe.
Copyright © 2020 Andermann, Torres Jiménez, Matos-Maraví, Batista, Blanco-Pastor, Gustafsson, Kistler, Liberal, Oxelman, Bacon and Antonelli.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources