

Life-Threatening Infectious Complications in Sickle Cell Disease: A Concise Narrative Review
- PMID: 32154192
- PMCID: PMC7044152
- DOI: 10.3389/fped.2020.00038
Life-Threatening Infectious Complications in Sickle Cell Disease: A Concise Narrative Review
Abstract
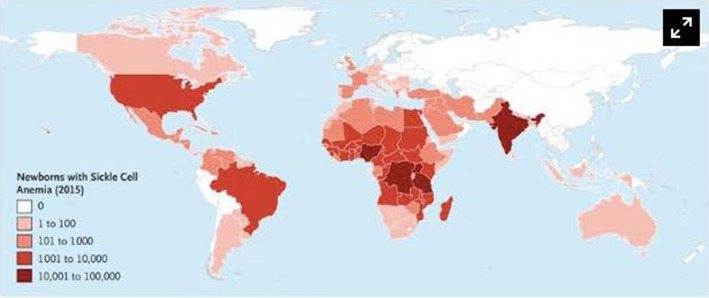
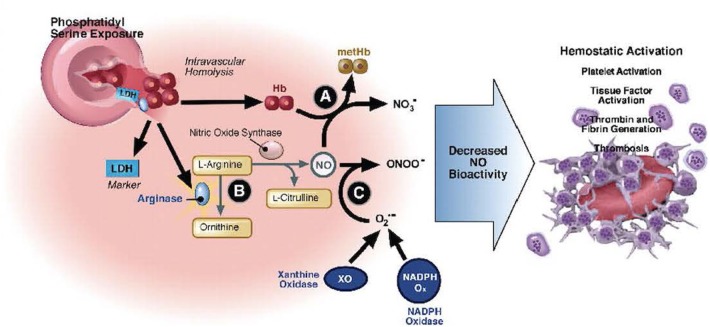
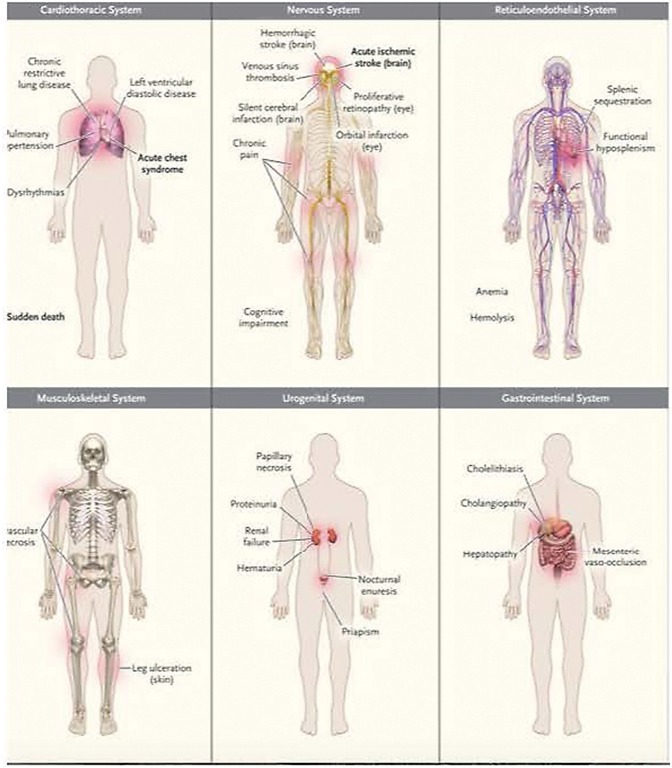
Sickle cell disease (SCD) results in chronic hemolytic anemia, recurrent vascular occlusion, insidious vital organ deterioration, early mortality, and diminished quality of life. Life-threatening acute physiologic crises may occur on a background of progressive diminishing vital organ function. Sickle hemoglobin polymerizes in the deoxygenated state, resulting in erythrocyte membrane deformation, vascular occlusion, and hemolysis. Vascular occlusion and increased blood viscosity results in functional asplenia and immune deficiency in early childhood, resulting in life-long increased susceptibility to serious bacterial infections. Infection remains a main cause of overall mortality in patients with SCD in low- and middle-income countries due to increased exposure to pathogens, increased co-morbidities such as malnutrition, lower vaccination rates, and diminished access to definitive care, including antibiotics and blood. Thus, the greatest gains in preventing infection-associated mortality can be achieved by addressing these factors for SCD patients in austere environments. In contrast, in high-income countries, perinatal diagnosis of SCD, antimicrobial prophylaxis, vaccination, aggressive use of antibiotics for febrile episodes, and the availability of contemporary critical care resources have resulted in a significant reduction in deaths from infection; however, chronic organ injury is problematic. All clinicians, regardless of their discipline, who assume the care of SCD patients must understand the importance of infectious disease as a contributor to death and disability. In this concise narrative review, we summarize the data that describes the importance of infectious diseases as a contributor to death and disability in SCD and discuss pathophysiology, prevalent organisms, prevention, management of acute episodes of critical illness, and ongoing care.
Keywords: children; critical care; infection; prophylaxis; sepsis; sickle cell disease; vaccination.
Copyright © 2020 Ochocinski, Dalal, Black, Carr, Lew, Sullivan and Kissoon.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources