

Splice variant of growth hormone-releasing hormone receptor drives esophageal squamous cell carcinoma conferring a therapeutic target
- PMID: 32156725
- PMCID: PMC7104313
- DOI: 10.1073/pnas.1913433117
Splice variant of growth hormone-releasing hormone receptor drives esophageal squamous cell carcinoma conferring a therapeutic target
Abstract
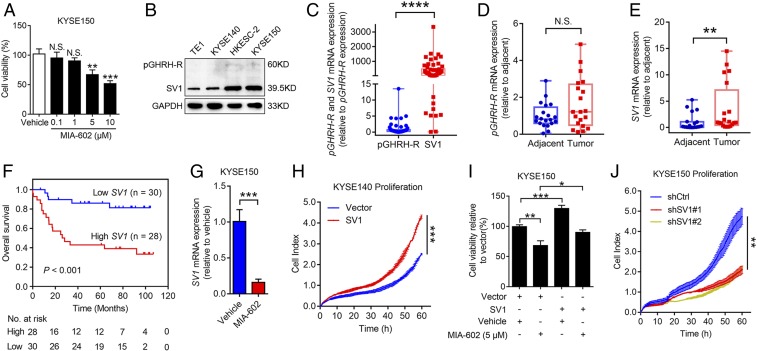
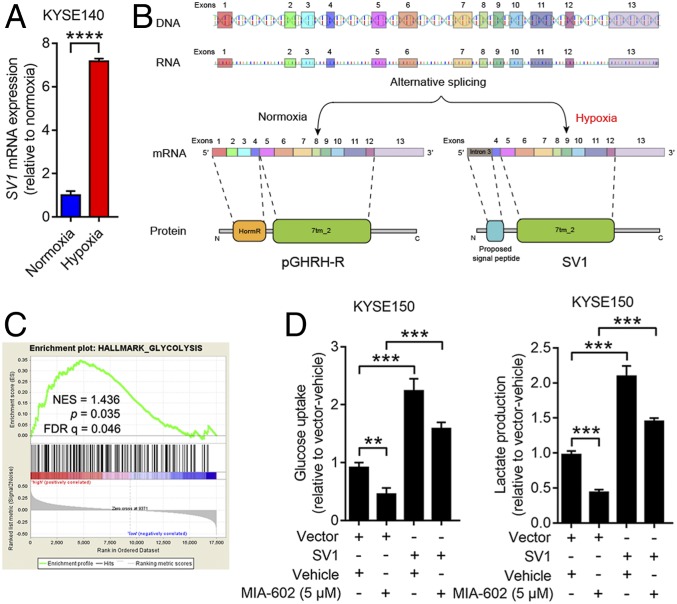
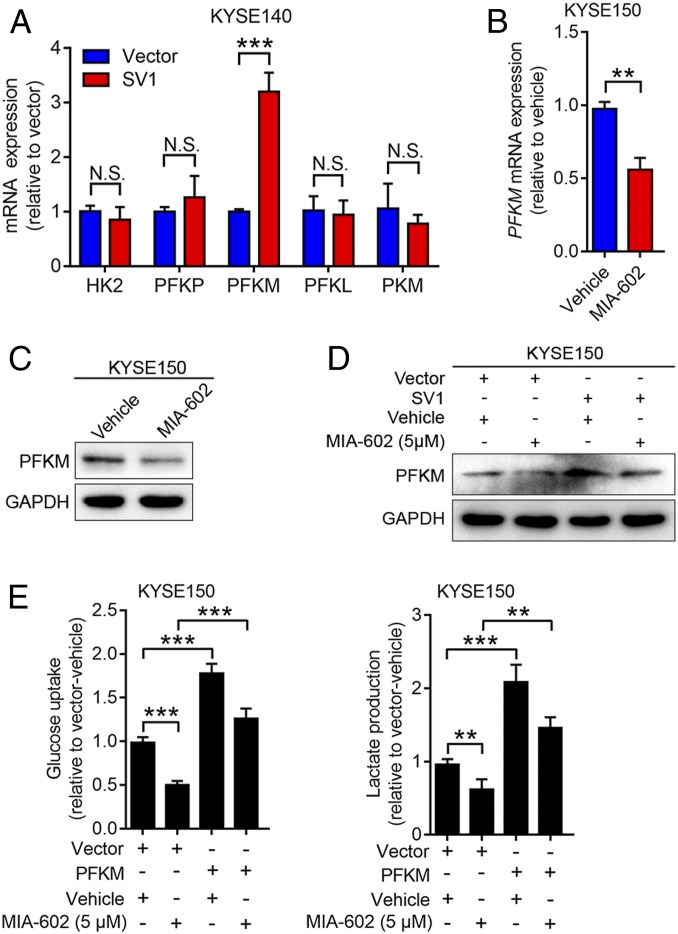
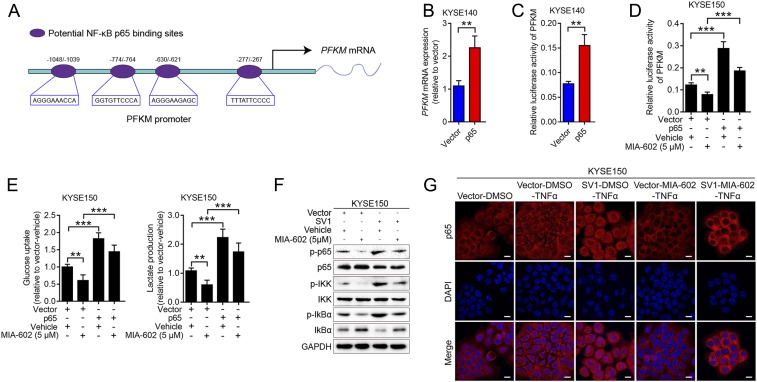
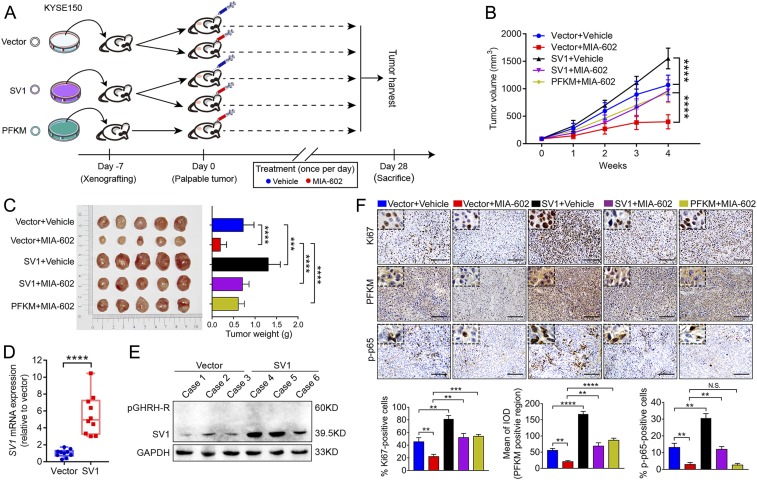
The extrahypothalamic growth hormone-releasing hormone (GHRH) and its cognate receptors (GHRH-Rs) and splice variants are expressed in a variety of cancers. It has been shown that the pituitary type of GHRH-R (pGHRH-R) mediates the inhibition of tumor growth induced by GHRH-R antagonists. However, GHRH-R antagonists can also suppress some cancers that do not express pGHRH-R, yet the underlying mechanisms have not been determined. Here, using human esophageal squamous cell carcinoma (ESCC) as a model, we were able to reveal that SV1, a known splice variant of GHRH-R, is responsible for the inhibition induced by GHRH-R antagonist MIA-602. We demonstrated that GHRH-R splice variant 1 (SV1) is a hypoxia-driven promoter of tumor progression. Hypoxia-elevated SV1 activates a key glycolytic enzyme, muscle-type phosphofructokinase (PFKM), through the nuclear factor kappa B (NF-κB) pathway, which enhances glycolytic metabolism and promotes progression of ESCC. The malignant actions induced by the SV1-NF-κB-PFKM pathway could be reversed by MIA-602. Altogether, our studies demonstrate a mechanism by which GHRH-R antagonists target SV1. Our findings suggest that SV1 is a hypoxia-induced oncogenic promoter which can be an alternative target of GHRH-R antagonists.
Keywords: GHRH-R antagonist; PFKM; glycolysis; hypoxia; splicing isoform of GHRH-R.
Copyright © 2020 the Author(s). Published by PNAS.
Conflict of interest statement
Competing interest statement: The Sponsor declares a conflict of interest. A.V.S. is a coinventor on the patent for growth hormone-releasing hormone analogs, assigned to the University of Miami, Miami, FL, and the Veterans Affairs Medical Center, Miami, FL. The other authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
