

Complementary Metagenomic Approaches Improve Reconstruction of Microbial Diversity in a Forest Soil
- PMID: 32156798
- PMCID: PMC7065516
- DOI: 10.1128/mSystems.00768-19
Complementary Metagenomic Approaches Improve Reconstruction of Microbial Diversity in a Forest Soil
Abstract
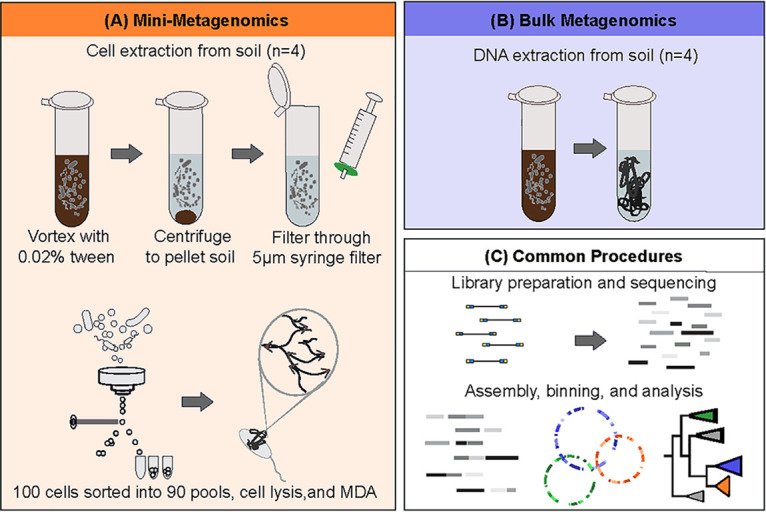
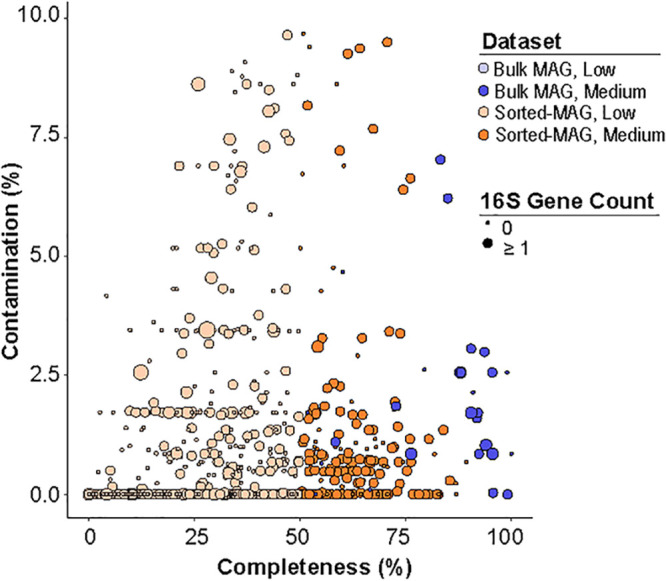
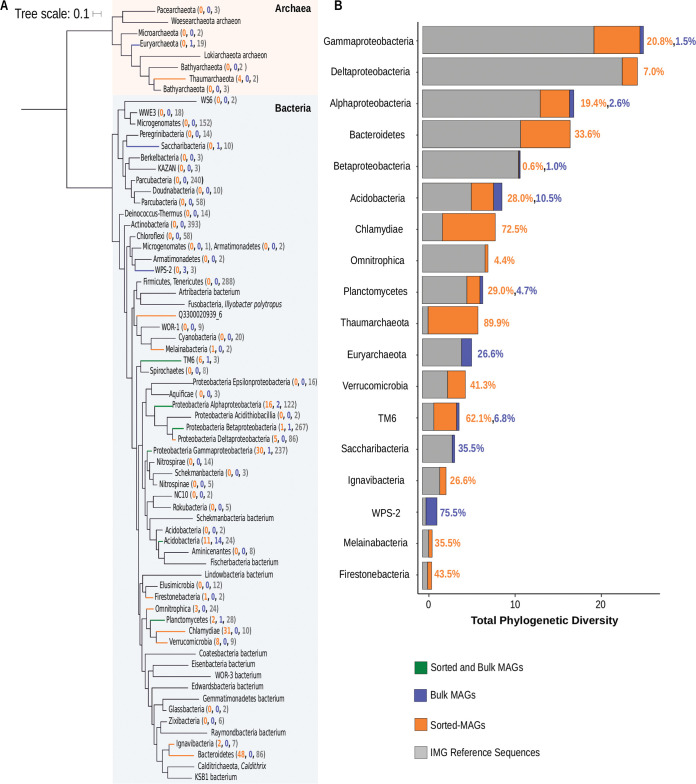
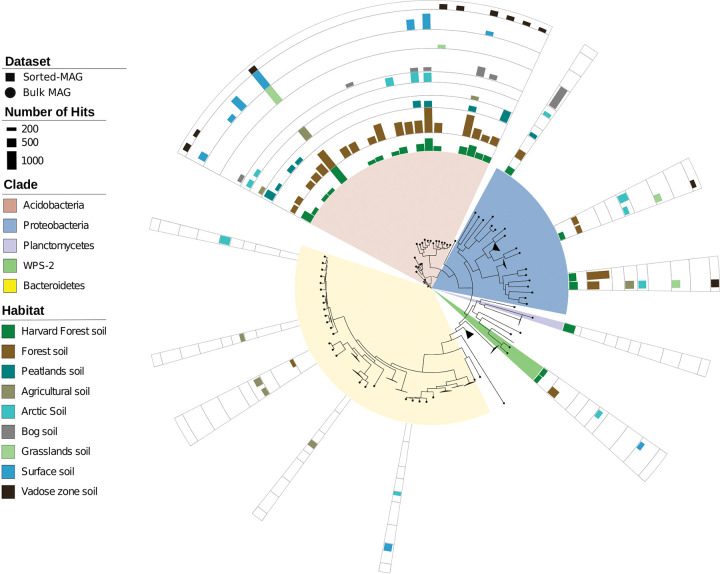
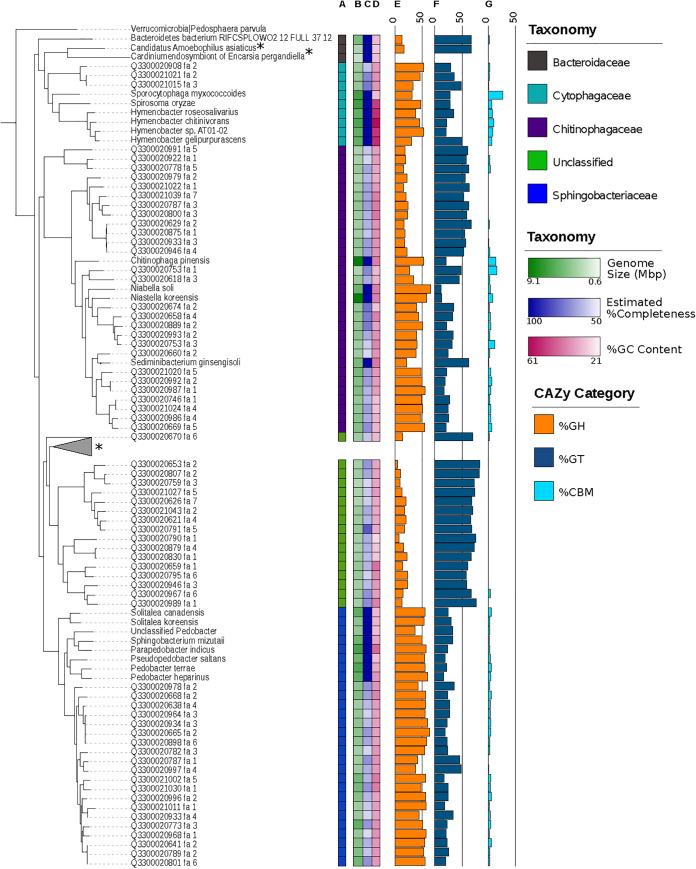
Soil ecosystems harbor diverse microorganisms and yet remain only partially characterized as neither single-cell sequencing nor whole-community sequencing offers a complete picture of these complex communities. Thus, the genetic and metabolic potential of this "uncultivated majority" remains underexplored. To address these challenges, we applied a pooled-cell-sorting-based mini-metagenomics approach and compared the results to bulk metagenomics. Informatic binning of these data produced 200 mini-metagenome assembled genomes (sorted-MAGs) and 29 bulk metagenome assembled genomes (MAGs). The sorted and bulk MAGs increased the known phylogenetic diversity of soil taxa by 7.2% with respect to the Joint Genome Institute IMG/M database and showed clade-specific sequence recruitment patterns across diverse terrestrial soil metagenomes. Additionally, sorted-MAGs expanded the rare biosphere not captured through MAGs from bulk sequences, exemplified through phylogenetic and functional analyses of members of the phylum Bacteroidetes Analysis of 67 Bacteroidetes sorted-MAGs showed conserved patterns of carbon metabolism across four clades. These results indicate that mini-metagenomics enables genome-resolved investigation of predicted metabolism and demonstrates the utility of combining metagenomics methods to tap into the diversity of heterogeneous microbial assemblages.IMPORTANCE Microbial ecologists have historically used cultivation-based approaches as well as amplicon sequencing and shotgun metagenomics to characterize microbial diversity in soil. However, challenges persist in the study of microbial diversity, including the recalcitrance of the majority of microorganisms to laboratory cultivation and limited sequence assembly from highly complex samples. The uncultivated majority thus remains a reservoir of untapped genetic diversity. To address some of the challenges associated with bulk metagenomics as well as low throughput of single-cell genomics, we applied flow cytometry-enabled mini-metagenomics to capture expanded microbial diversity from forest soil and compare it to soil bulk metagenomics. Our resulting data from this pooled-cell sorting approach combined with bulk metagenomics revealed increased phylogenetic diversity through novel soil taxa and rare biosphere members. In-depth analysis of genomes within the highly represented Bacteroidetes phylum provided insights into conserved and clade-specific patterns of carbon metabolism.
Keywords: flow cytometry; metagenomics; microbial ecology; soil microbiology.
Copyright © 2020 Alteio et al.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
