

Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes
- PMID: 32157095
- PMCID: PMC7064602
- DOI: 10.1038/s41467-020-14968-9
Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes
Abstract
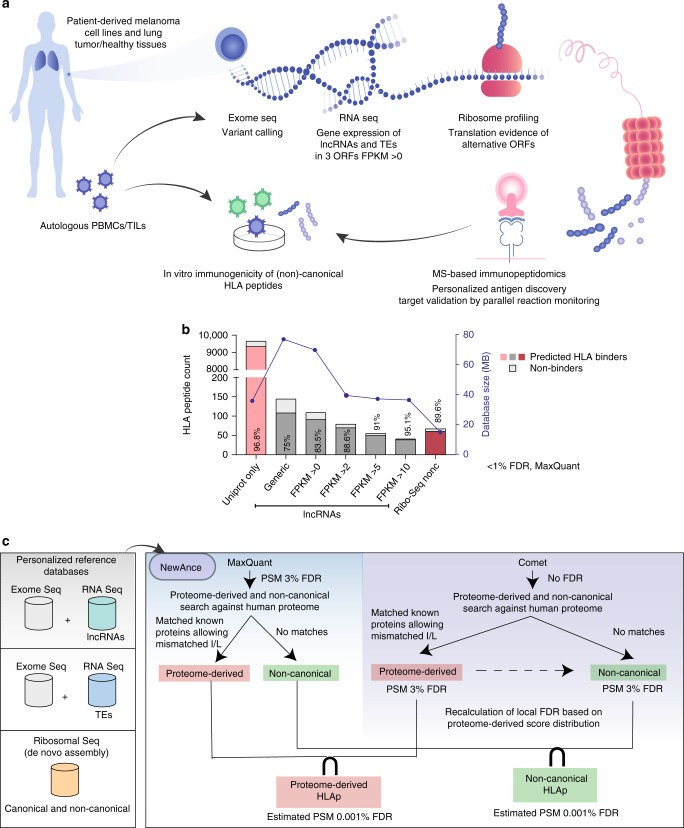
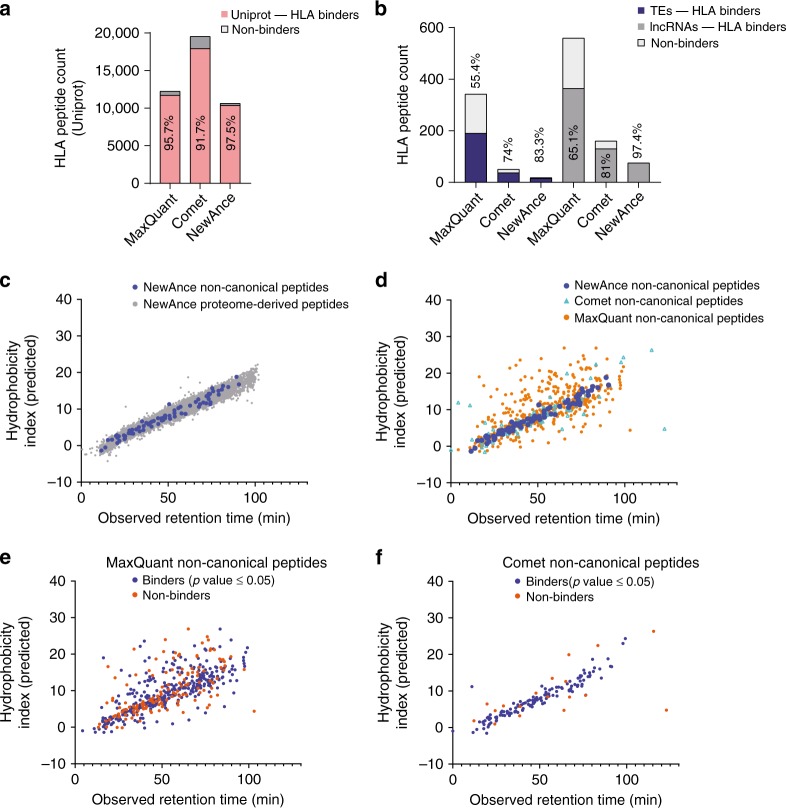
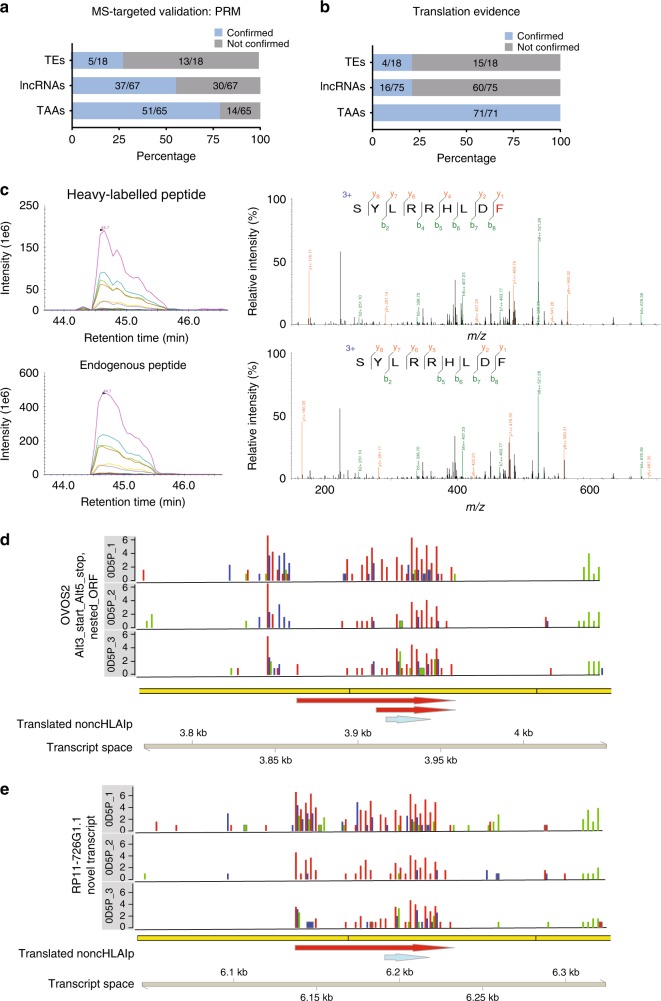
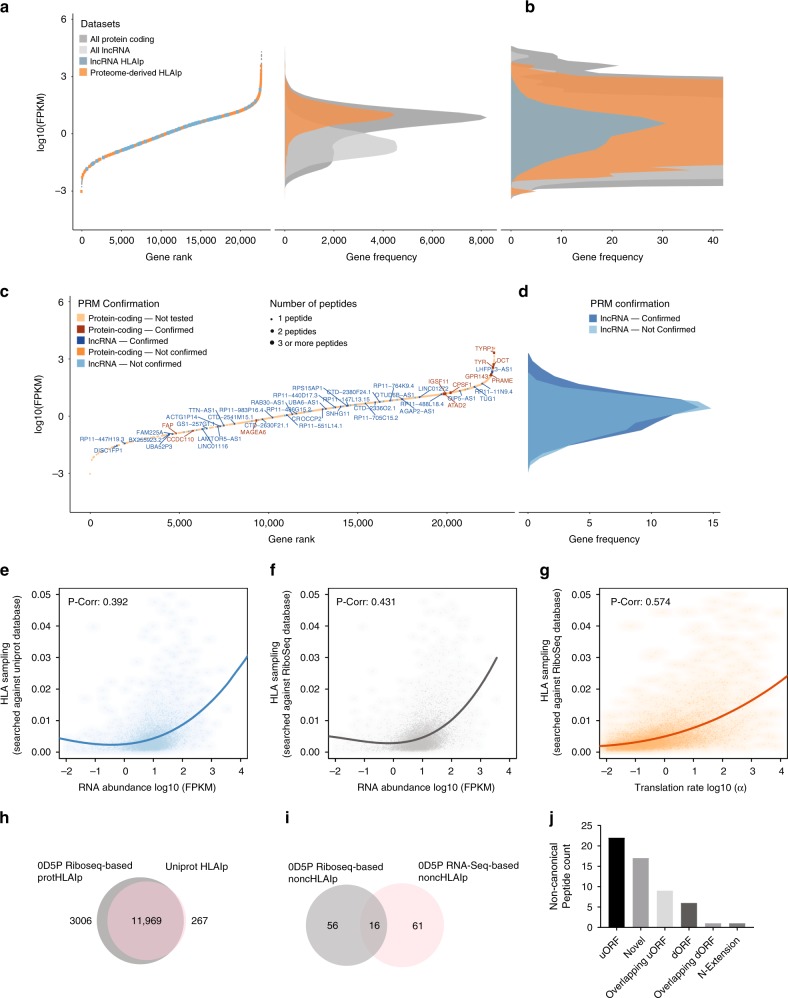
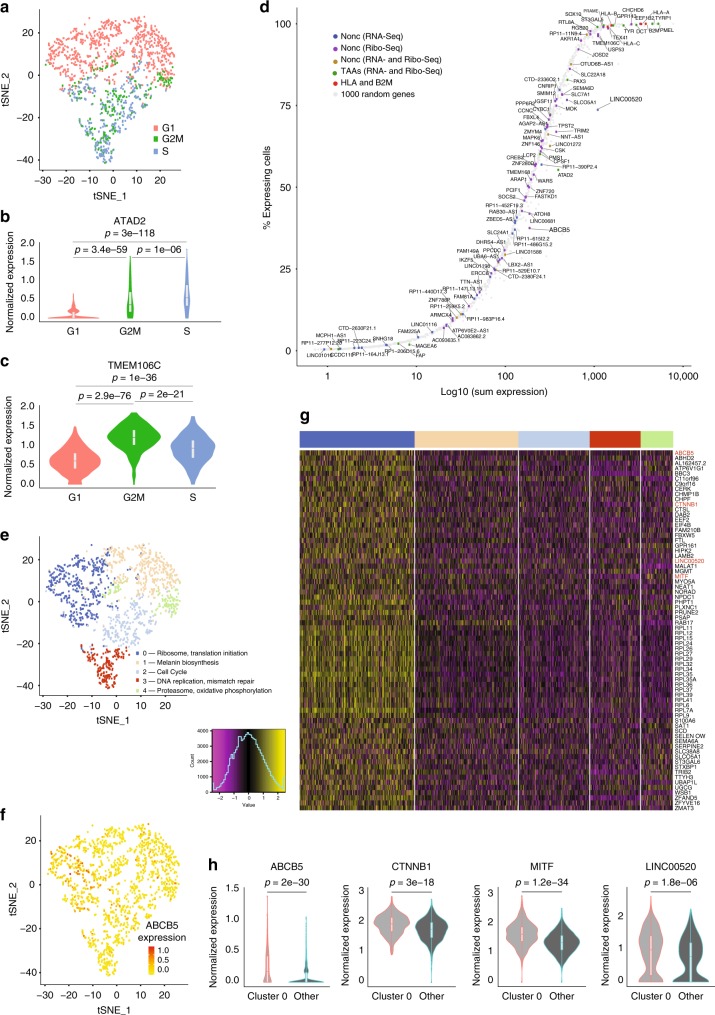
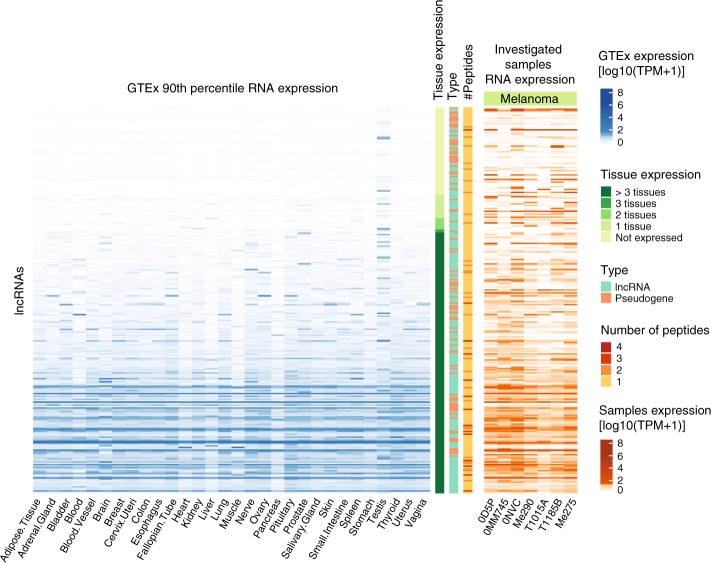
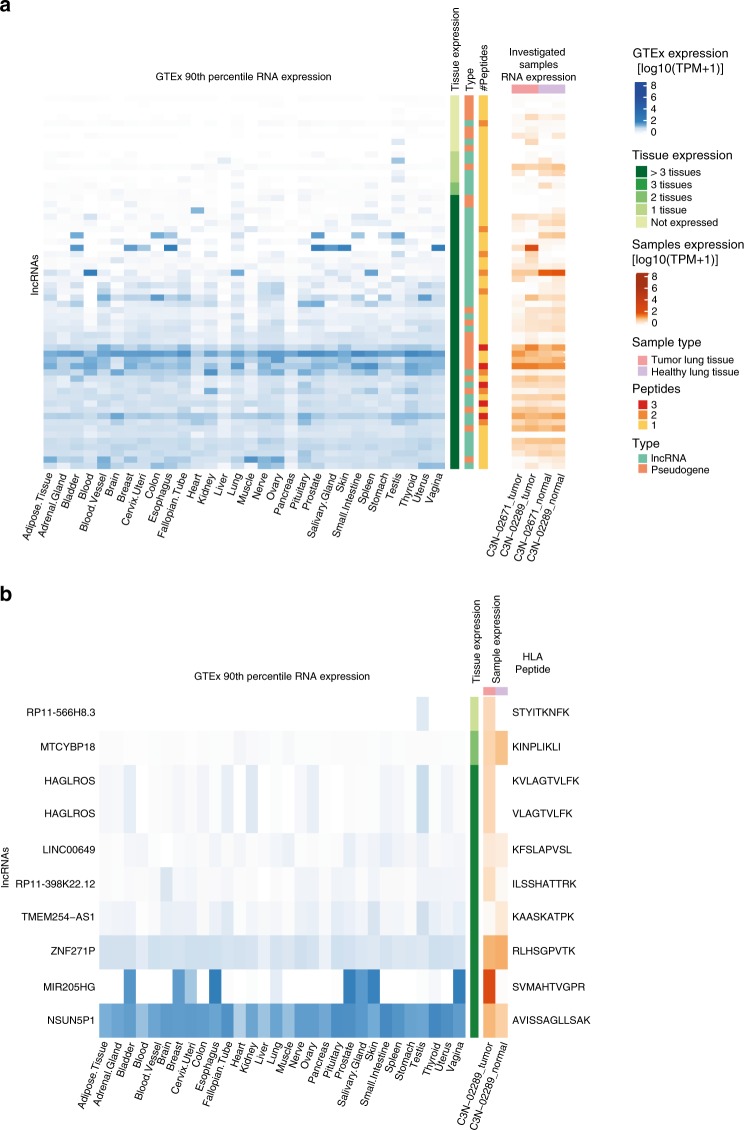
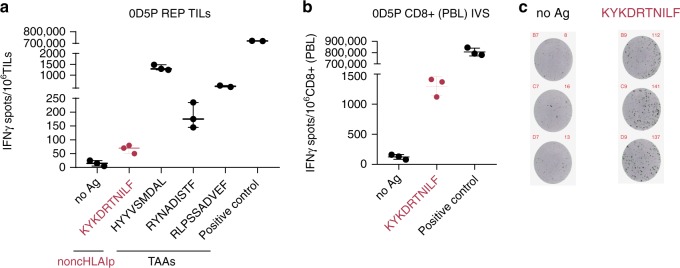
Efforts to precisely identify tumor human leukocyte antigen (HLA) bound peptides capable of mediating T cell-based tumor rejection still face important challenges. Recent studies suggest that non-canonical tumor-specific HLA peptides derived from annotated non-coding regions could elicit anti-tumor immune responses. However, sensitive and accurate mass spectrometry (MS)-based proteogenomics approaches are required to robustly identify these non-canonical peptides. We present an MS-based analytical approach that characterizes the non-canonical tumor HLA peptide repertoire, by incorporating whole exome sequencing, bulk and single-cell transcriptomics, ribosome profiling, and two MS/MS search tools in combination. This approach results in the accurate identification of hundreds of shared and tumor-specific non-canonical HLA peptides, including an immunogenic peptide derived from an open reading frame downstream of the melanoma stem cell marker gene ABCB5. These findings hold great promise for the discovery of previously unknown tumor antigens for cancer immunotherapy.
Conflict of interest statement
G.C. has received grants and research support from BMS, Celgene, Boehringer Ingelheim, Roche, Lovance and Kite, and worked with them as a coinvestigator in clinical trials. G.C. has received honouraria for consultations or presentations from Roche, Genentech, BMS, AstraZeneca, Sanofi-Aventis, Nextcure, and GeneosTx. G.C. has patents regarding antibodies and vaccines targeting the tumor vasculature as well as technologies related to T cell expansion and engineering for T cell therapy. G.C. receives royalties from the University of Pennsylvania. The remaining authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
