

Identification of a novel gene signature for the prediction of recurrence in HCC patients by machine learning of genome-wide databases
- PMID: 32157118
- PMCID: PMC7064516
- DOI: 10.1038/s41598-020-61298-3
Identification of a novel gene signature for the prediction of recurrence in HCC patients by machine learning of genome-wide databases
Abstract
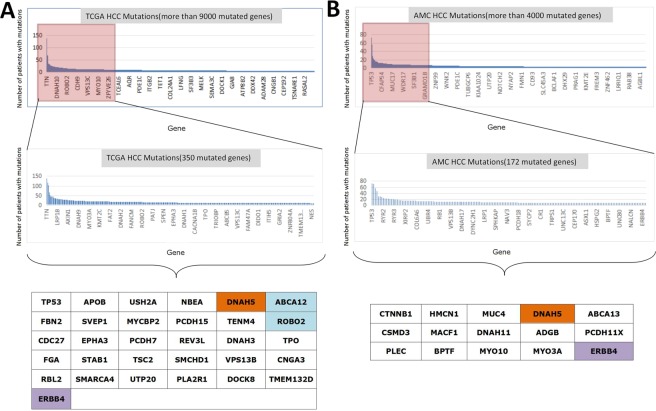
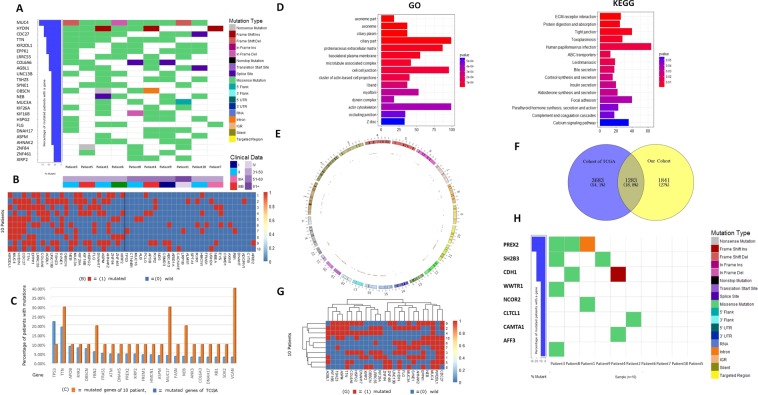
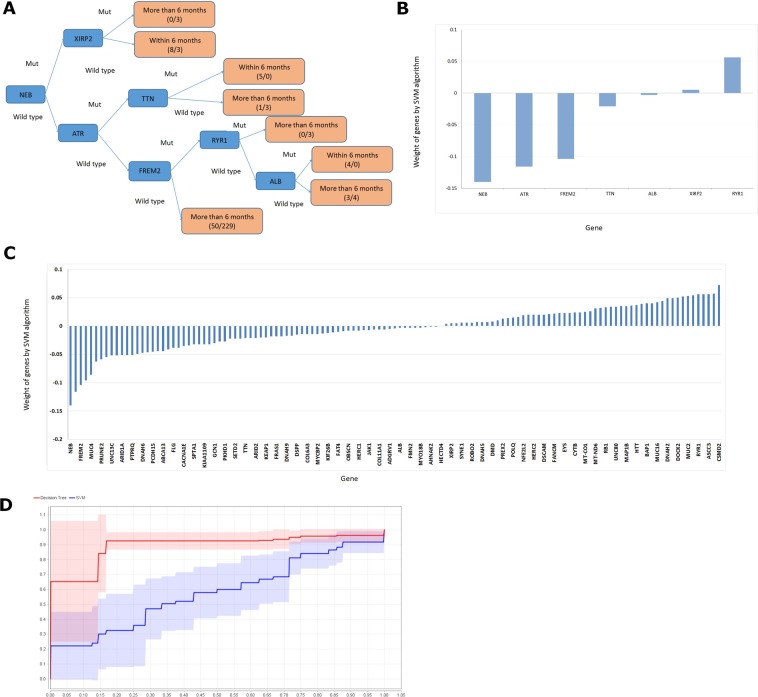
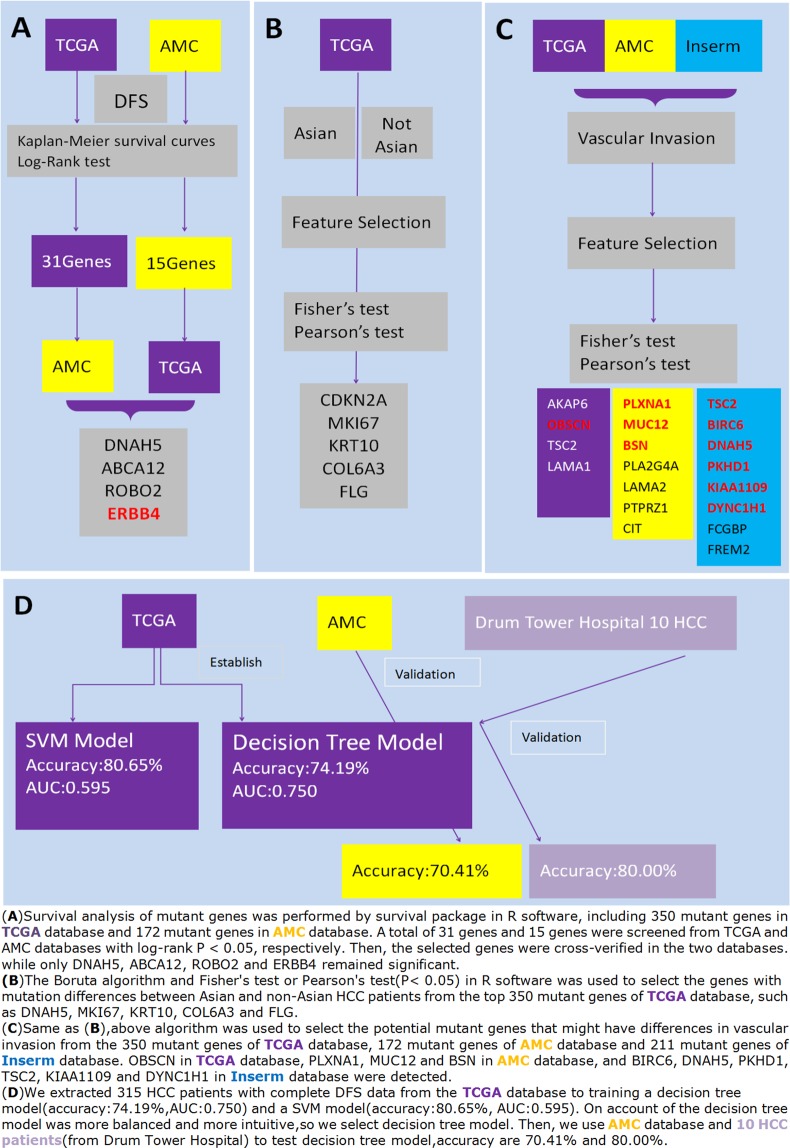
Hepatocellular carcinoma (HCC) is a common malignant tumor in China. In the present study, we aimed to construct and verify a prediction model of recurrence in HCC patients using databases (TCGA, AMC and Inserm) and machine learning methods and obtain the gene signature that could predict early relapse of HCC. Statistical methods, such as feature selection, survival analysis and Chi-Square test in R software, were used to analyze and select mutant genes related to disease free survival (DFS), race and vascular invasion. In addition, whole-exome sequencing was performed on 10 HCC patients recruited from our center, and the sequencing results were compared with the databases. Using the databases and machine learning methods, the prediction model of recurrence was constructed and optimized, and the selected mutant genes were verified in the test group. The accuracy of prediction was 74.19%. Moreover, these 10 patients from our center were used to verify these mutant genes and the prediction model, and a success rate of 80% was achieved. Collectively, we discovered recurrence-related genes and established recurrence prediction model of recurrence for HCC patients, which could provide significant guidance for clinical prediction of recurrence.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Six-long non-coding RNA signature predicts recurrence-free survival in hepatocellular carcinoma.World J Gastroenterol. 2019 Jan 14;25(2):220-232. doi: 10.3748/wjg.v25.i2.220. World J Gastroenterol. 2019. PMID: 30670911 Free PMC article.
-
A gene-based risk score model for predicting recurrence-free survival in patients with hepatocellular carcinoma.BMC Cancer. 2021 Jan 5;21(1):6. doi: 10.1186/s12885-020-07692-6. BMC Cancer. 2021. PMID: 33402113 Free PMC article.
-
A six-gene-based prognostic signature for hepatocellular carcinoma overall survival prediction.Life Sci. 2018 Jun 15;203:83-91. doi: 10.1016/j.lfs.2018.04.025. Epub 2018 Apr 17. Life Sci. 2018. PMID: 29678742
-
Identification and validation of a potent multi-mRNA signature for the prediction of early relapse in hepatocellular carcinoma.Carcinogenesis. 2019 Jul 20;40(7):840-852. doi: 10.1093/carcin/bgz018. Carcinogenesis. 2019. PMID: 31059567
-
Mining Prognostic Biomarkers of Hepatocellular Carcinoma Based on Immune-Associated Genes.DNA Cell Biol. 2020 Apr;39(4):499-512. doi: 10.1089/dna.2019.5099. Epub 2020 Feb 18. DNA Cell Biol. 2020. PMID: 32069130 Review.
Cited by
-
Combining a machine-learning derived 4-lncRNA signature with AFP and TNM stages in predicting early recurrence of hepatocellular carcinoma.BMC Genomics. 2023 Feb 27;24(1):89. doi: 10.1186/s12864-023-09194-8. BMC Genomics. 2023. PMID: 36849926 Free PMC article.
-
Enhancing selection of alcohol consumption-associated genes by random forest.Br J Nutr. 2024 Jun 28;131(12):2058-2067. doi: 10.1017/S0007114524000795. Epub 2024 Apr 12. Br J Nutr. 2024. PMID: 38606596 Free PMC article.
-
The Role of Artificial Intelligence in the Detection and Implementation of Biomarkers for Hepatocellular Carcinoma: Outlook and Opportunities.Cancers (Basel). 2023 May 26;15(11):2928. doi: 10.3390/cancers15112928. Cancers (Basel). 2023. PMID: 37296890 Free PMC article. Review.
-
Role of three-dimensional printing and artificial intelligence in the management of hepatocellular carcinoma: Challenges and opportunities.World J Gastrointest Oncol. 2022 Apr 15;14(4):765-793. doi: 10.4251/wjgo.v14.i4.765. World J Gastrointest Oncol. 2022. PMID: 35582107 Free PMC article. Review.
-
A novel classification algorithm for customer churn prediction based on hybrid Ensemble-Fusion model.Sci Rep. 2024 Aug 30;14(1):20179. doi: 10.1038/s41598-024-71168-x. Sci Rep. 2024. PMID: 39215049 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
