

Adjusting for age improves identification of gut microbiome alterations in multiple diseases
- PMID: 32159510
- PMCID: PMC7065848
- DOI: 10.7554/eLife.50240
Adjusting for age improves identification of gut microbiome alterations in multiple diseases
Abstract
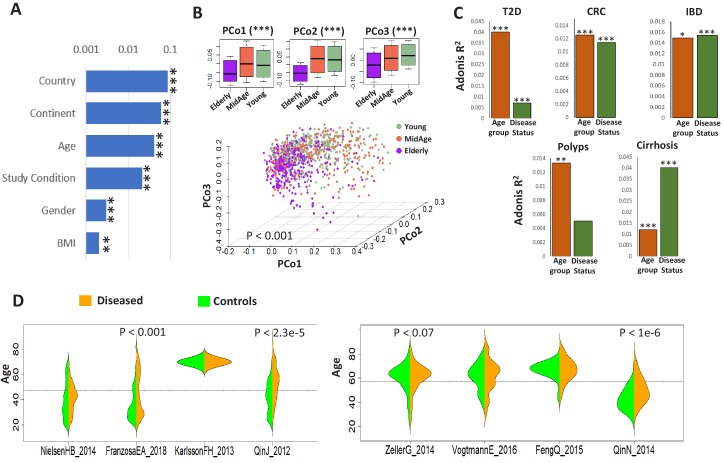
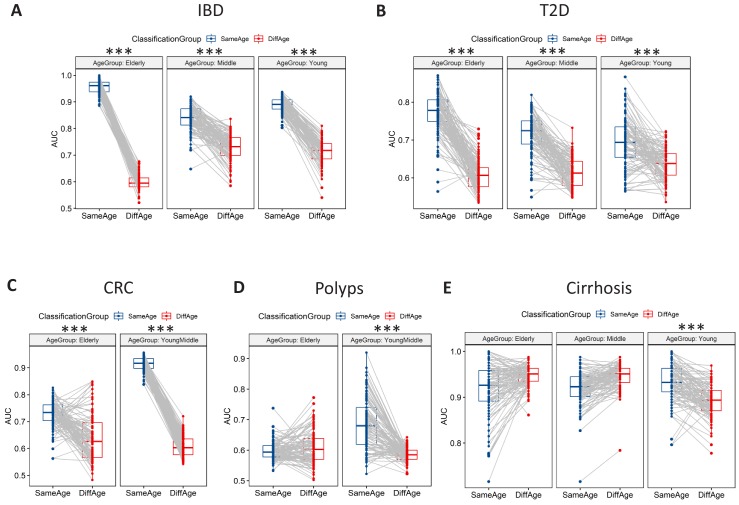
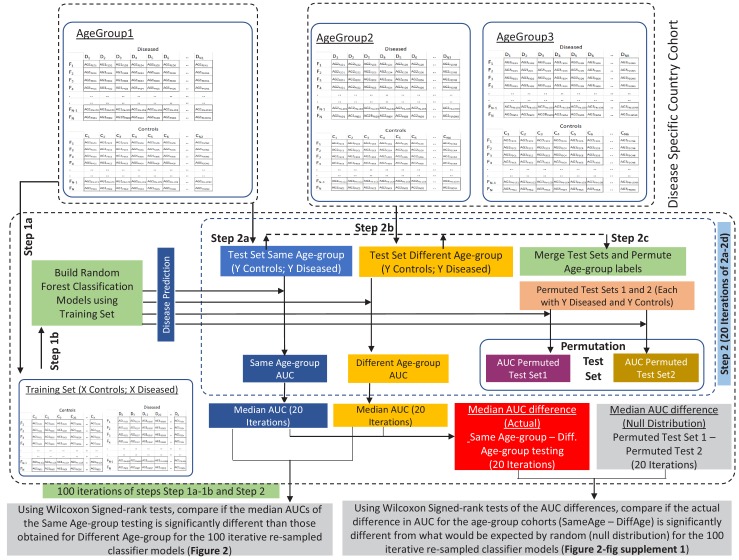
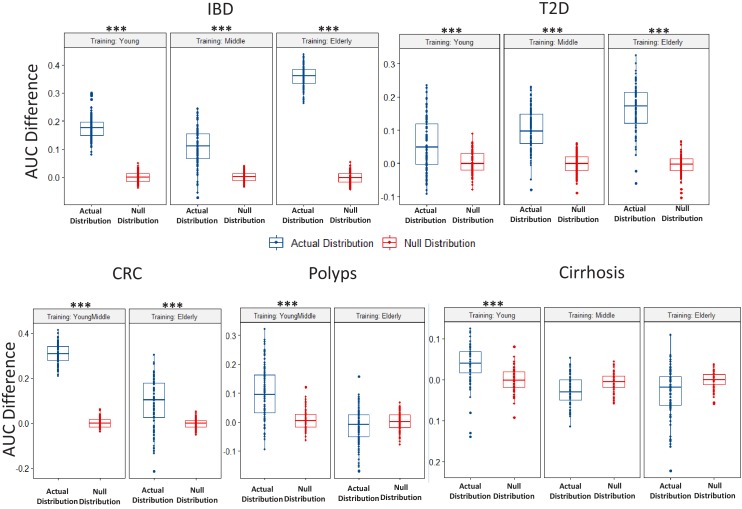
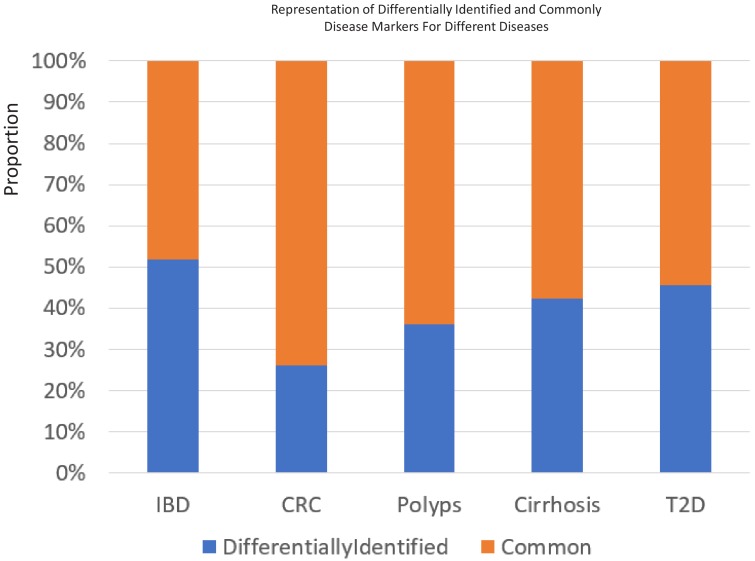
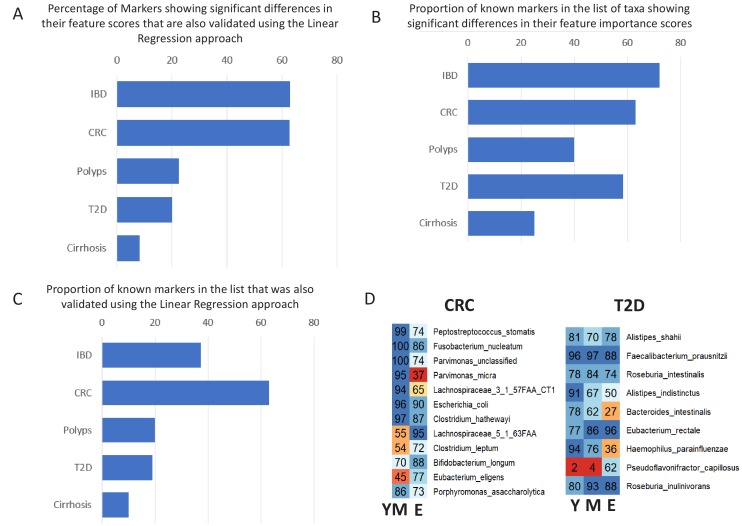
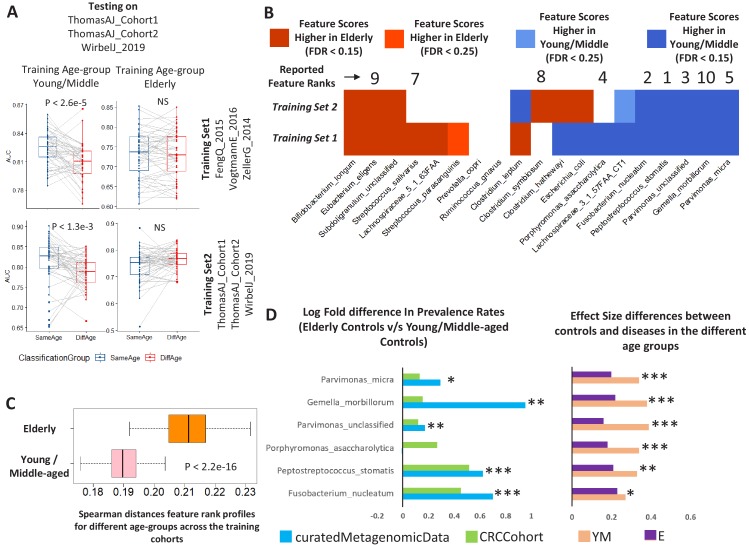
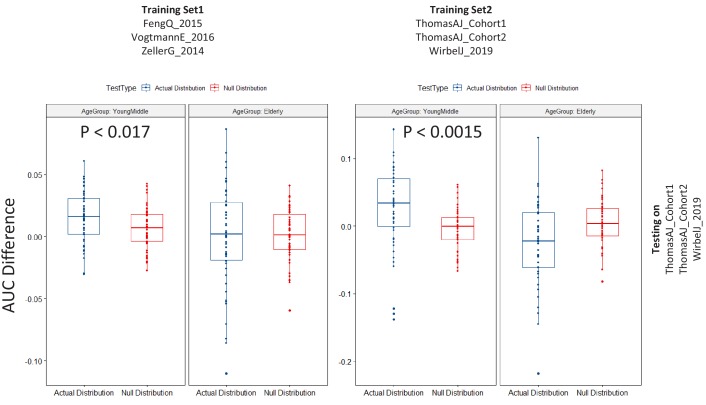
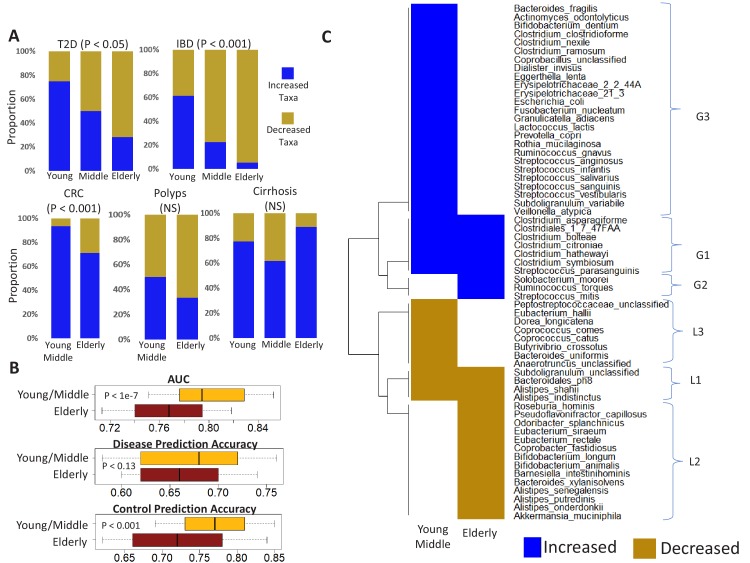
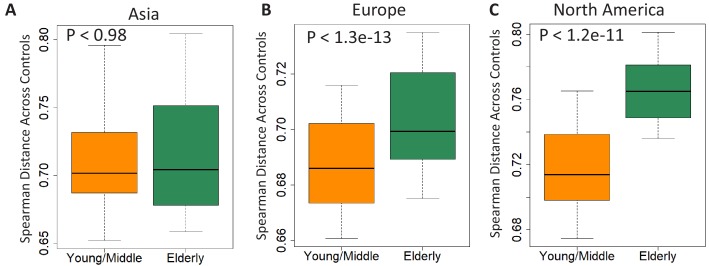
Interaction between disease-microbiome associations and ageing has not been explored in detail. Here, using age/region-matched sub-sets, we analysed the gut microbiome differences across five major diseases in a multi-cohort dataset constituting more than 2500 individuals from 20 to 89 years old. We show that disease-microbiome associations display specific age-centric trends. Ageing-associated microbiome alterations towards a disease-like configuration occur in colorectal cancer patients, thereby masking disease signatures. We identified a microbiome disease response shared across multiple diseases in elderly subjects that is distinct from that in young/middle-aged individuals, but also a novel set of taxa consistently gained in disease across all age groups. A subset of these taxa was associated with increased frailty in subjects from the ELDERMET cohort. The relevant taxa differentially encode specific functions that are known to have disease associations.
Keywords: ageing; computational biology; disease-microbiome; host-microbiome; infectious disease; microbiology; systems biology.
Plain language summary
The human body is an ecosystem made up of both human cells and trillions of microbes, and the largest microbial community is in the gut. This community of gut microbes helps harvest nutrients from our food, modulates our immune system, and even affects our mood. Infectious and chronic diseases appear to cause changes in the make-up of the gut microbiome, while microbiome changes may increase the risk of some non-infectious diseases. Learning more about these disease-linked changes in the gut microbiome may therefore help scientists to develop new tests and treatments. To do this, scientists need to understand which microbes play a role in individual diseases, if risk-related microbes are gained or helpful microbes lost in patients with particular diseases, and if certain changes in gut microbes occur across many diseases. Ageing also changes the gut microbes. This may happen because older individuals eat a less complex diet and are likely to take many medications that may alter the microbes in their gut. Because of this, age may affect changes in gut microbes associated with diseases. This highlights the need for studies that tease apart the importance of ageing-related and disease-related changes in the gut microbiome. Now, Ghosh et al. show that gut microbe changes linked to diseases may vary with a person’s age. The analysis compared the gut microbiomes of more than 2,500 individuals aged 20 to 89. This included individuals with inflammatory bowel disease, colorectal cancer, type 2 diabetes, intestinal polyps and liver cirrhosis. The study revealed that younger people gradually gain disease-associated gut microbes, while older people tend to lose the gut microbes usually found in a healthy gut. Ghosh et al. also identified a set of gut microbes that were gained in many diseases and across age-groups. This set of microbes was also associated with frailty in elderly people. The characteristics of the microbes in this set are all known to have detrimental effects on human health. This analysis shows how important it is to control for age and other factors that may skew the results of microbiome projects. Future studies are needed to understand why these gut microbe changes occur and what the consequences of these changes are for a person’s health and the course of their disease. This may lead to the development of treatment strategies that help promote a healthy gut microbiome and fight disease throughout life.
© 2020, Ghosh et al.
Conflict of interest statement
TG, MD, IJ, PO No competing interests declared
Figures









References
-
- Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, Littmann E, van den Brink MR, Jenq RR, Taur Y, Sander C, Cross JR, Toussaint NC, Xavier JB, Pamer EG. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature. 2015;517:205–208. doi: 10.1038/nature13828. - DOI - PMC - PubMed
-
- Claesson MJ, Jeffery IB, Conde S, Power SE, O'Connor EM, Cusack S, Harris HM, Coakley M, Lakshminarayanan B, O'Sullivan O, Fitzgerald GF, Deane J, O'Connor M, Harnedy N, O'Connor K, O'Mahony D, van Sinderen D, Wallace M, Brennan L, Stanton C, Marchesi JR, Fitzgerald AP, Shanahan F, Hill C, Ross RP, O'Toole PW. Gut Microbiota composition correlates with diet and health in the elderly. Nature. 2012;488:178–184. doi: 10.1038/nature11319. - DOI - PubMed
-
- Deschasaux M, Bouter KE, Prodan A, Levin E, Groen AK, Herrema H, Tremaroli V, Bakker GJ, Attaye I, Pinto-Sietsma SJ, van Raalte DH, Snijder MB, Nicolaou M, Peters R, Zwinderman AH, Bäckhed F, Nieuwdorp M. Depicting the composition of gut Microbiota in a population with varied ethnic origins but shared geography. Nature Medicine. 2018;24:1526–1531. doi: 10.1038/s41591-018-0160-1. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
