

Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds
- PMID: 32162456
- PMCID: PMC7228259
- DOI: 10.1002/minf.202000028
Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds
Abstract
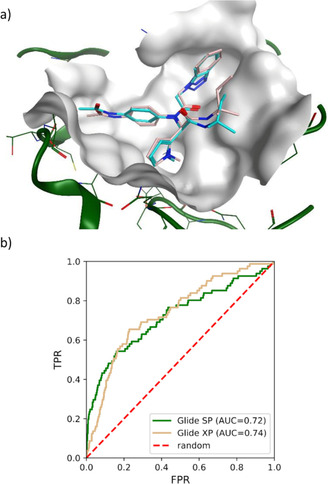
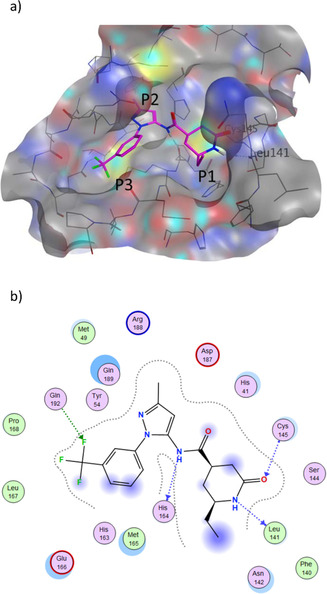
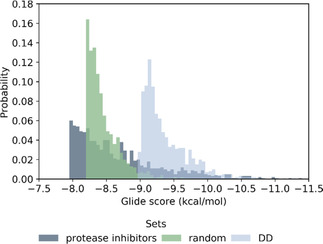
The recently emerged 2019 Novel Coronavirus (SARS-CoV-2) and associated COVID-19 disease cause serious or even fatal respiratory tract infection and yet no approved therapeutics or effective treatment is currently available to effectively combat the outbreak. This urgent situation is pressing the world to respond with the development of novel vaccine or a small molecule therapeutics for SARS-CoV-2. Along these efforts, the structure of SARS-CoV-2 main protease (Mpro) has been rapidly resolved and made publicly available to facilitate global efforts to develop novel drug candidates. Recently, our group has developed a novel deep learning platform - Deep Docking (DD) which provides fast prediction of docking scores of Glide (or any other docking program) and, hence, enables structure-based virtual screening of billions of purchasable molecules in a short time. In the current study we applied DD to all 1.3 billion compounds from ZINC15 library to identify top 1,000 potential ligands for SARS-CoV-2 Mpro protein. The compounds are made publicly available for further characterization and development by scientific community.
Keywords: COVID-19; SARS-CoV-2; deep learning; protease inhibitors; virtual screening.
© 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
Conflict of interest statement
None declared.
Figures

References
-
- “Coronavirus latest: Chinese cases spike after changes to diagnosis method,” can be found under http://www.nature.com/articles/d41586-020-00154-w, 2020.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
