

Molecular and clinical studies in 107 Noonan syndrome affected individuals with PTPN11 mutations
- PMID: 32164556
- PMCID: PMC7068896
- DOI: 10.1186/s12881-020-0986-5
Molecular and clinical studies in 107 Noonan syndrome affected individuals with PTPN11 mutations
Abstract
Background: Noonan syndrome (NS), an autosomal dominant developmental genetic disorder, is caused by germline mutations in genes associated with the RAS / mitogen-activated protein kinase (MAPK) pathway. In several studies PTPN11 is one of the genes with a significant number of pathogenic variants in NS-affected patients. Therefore, clinically diagnosed NS individuals are initially tested for pathogenic variants in PTPN11 gene to confirm the relationship before studying genotype-phenotype correlation.
Methods: Individuals (363) with clinically diagnosed NS from four hospitals in South India were recruited and the exons of PTPN11 gene were sequenced.
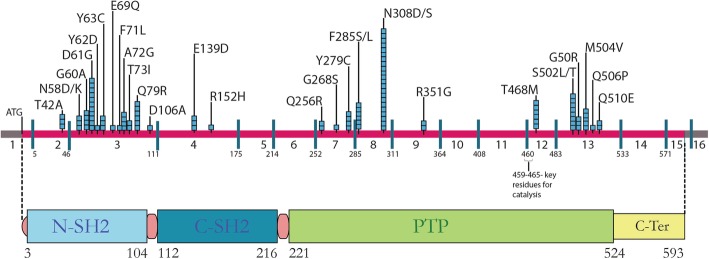
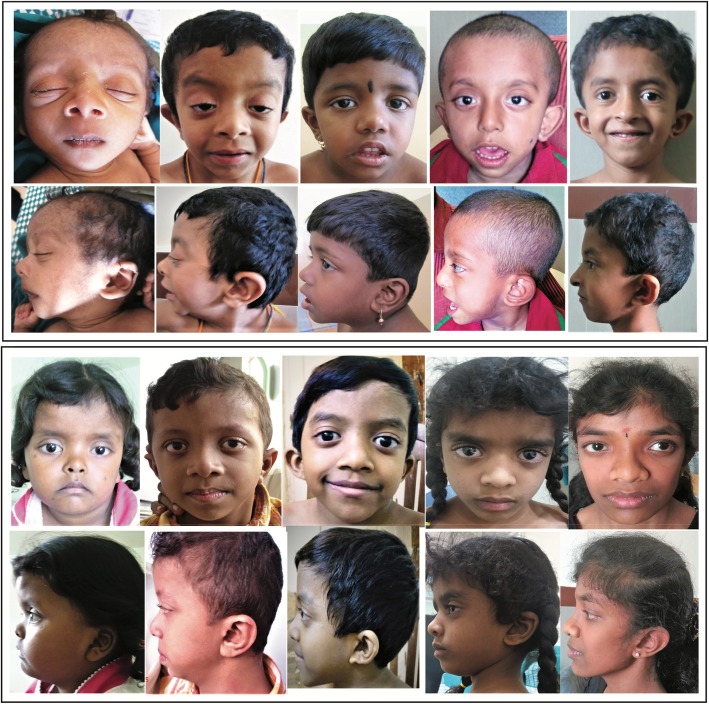
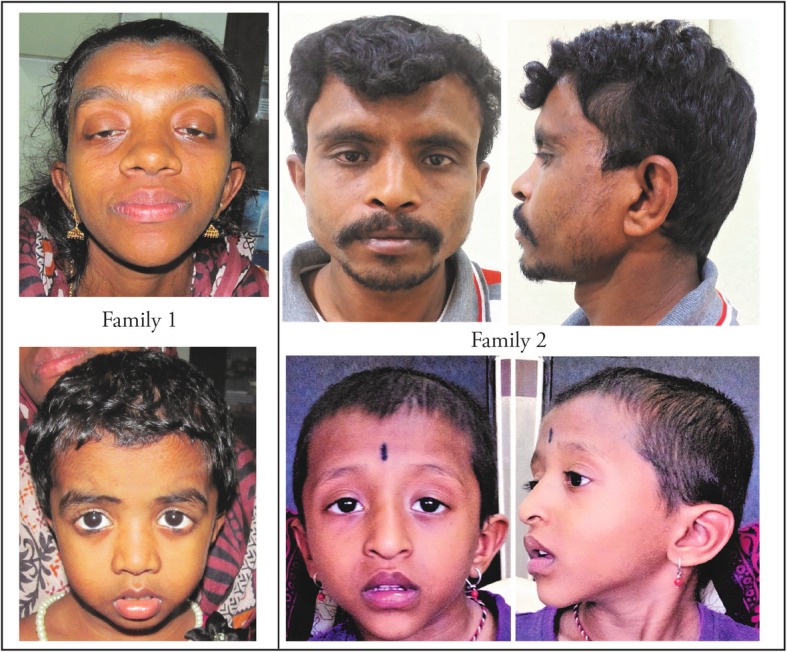
Results: Thirty-two previously described pathogenic variants in eight different exons in PTPN11 gene were detected in 107 patients, of whom 10 were familial cases. Exons 3, 8 and 13 had the highest number of pathogenic variants. The most commonly identified pathogenic variants in this series were in exon 8 (c.922A > G, c.923A > G), observed in 22 of the affected. Congenital cardiac anomalies were present in 84% of the mutation-positive cohort, the majority being defects in the right side of the heart. The most common facial features were downward-slanting palpebral fissures, hypertelorism and low-set posteriorly rotated ears. Other clinical features included short stature (40%), pectus excavatum (54%) and, in males, unilateral or bilateral cryptorchidism (44%).
Conclusion: The clinical features and mutational spectrum observed in our cohort are similar to those reported in other large studies done worldwide. This is the largest case series of NS-affected individuals with PTPN11 mutations described till date from India.
Keywords: Congenital heart defects; Mutational analysis; Noonan syndrome; PTPN11; RASopathy; SHP-2.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Cesur Aydin K, Ozcan I, Bona G. Noonan syndrome: A review. Minerva Pediatr. 2008;60:343–346. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
