

mtDNA Activates cGAS Signaling and Suppresses the YAP-Mediated Endothelial Cell Proliferation Program to Promote Inflammatory Injury
- PMID: 32164878
- PMCID: PMC7266657
- DOI: 10.1016/j.immuni.2020.02.002
mtDNA Activates cGAS Signaling and Suppresses the YAP-Mediated Endothelial Cell Proliferation Program to Promote Inflammatory Injury
Abstract
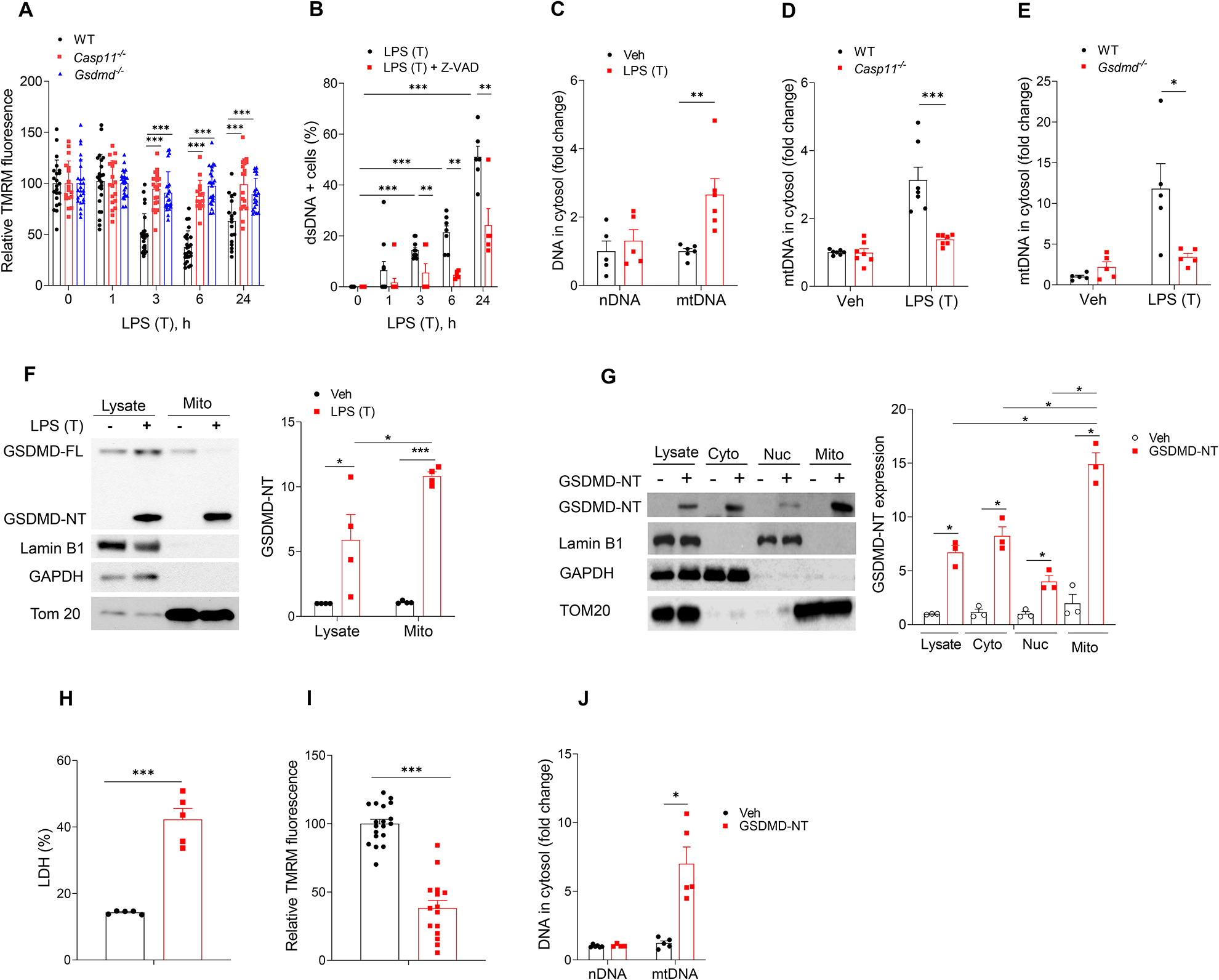
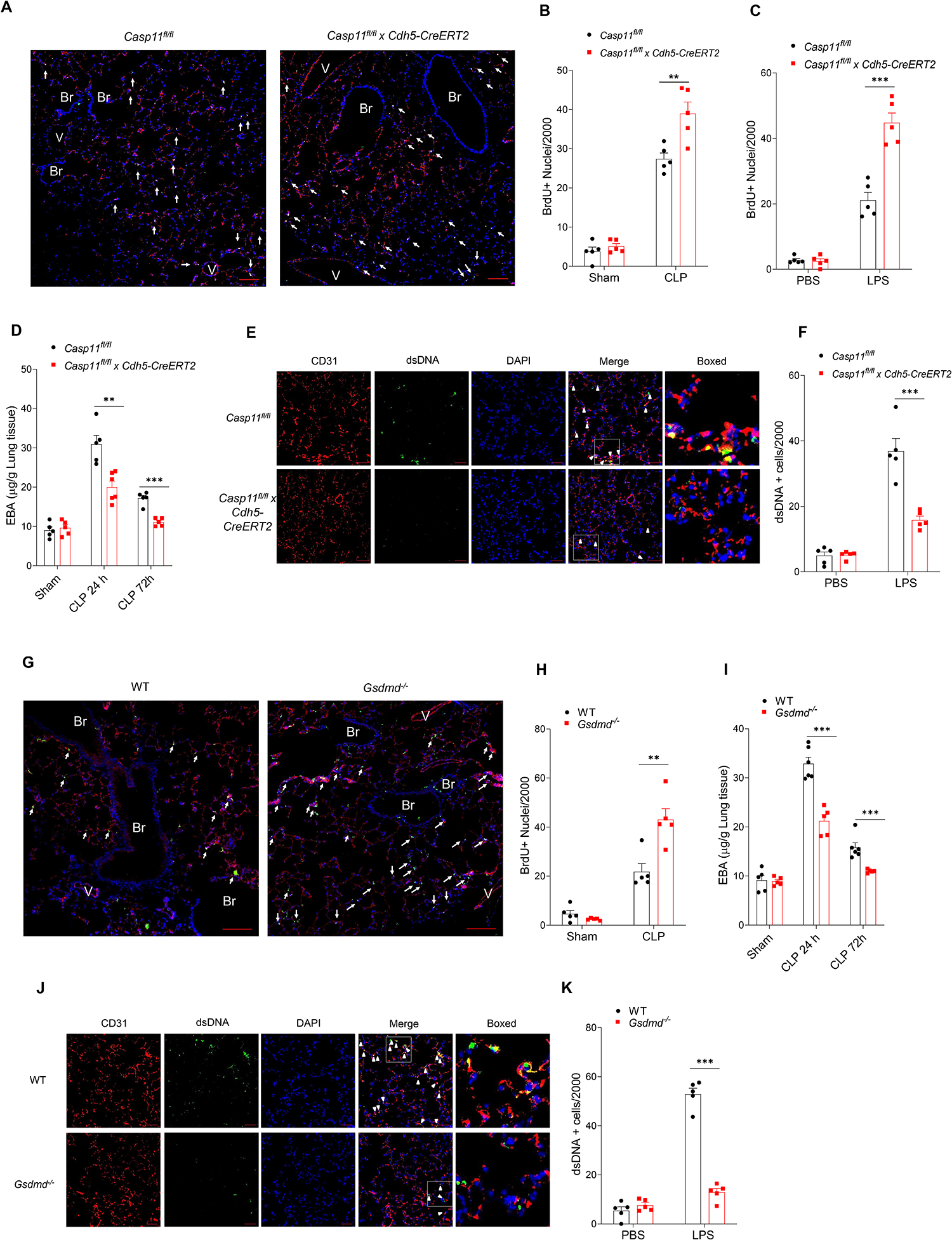
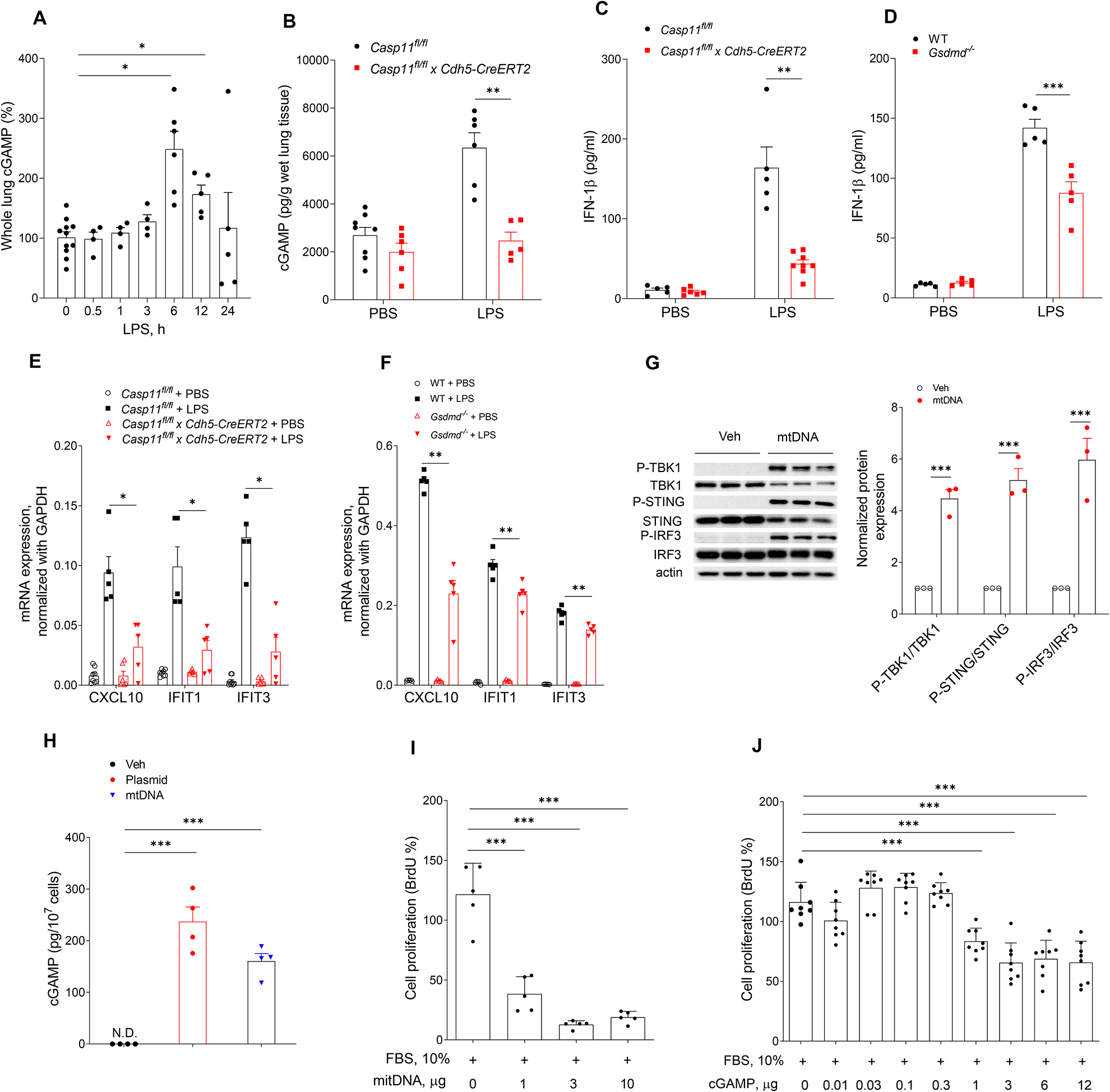
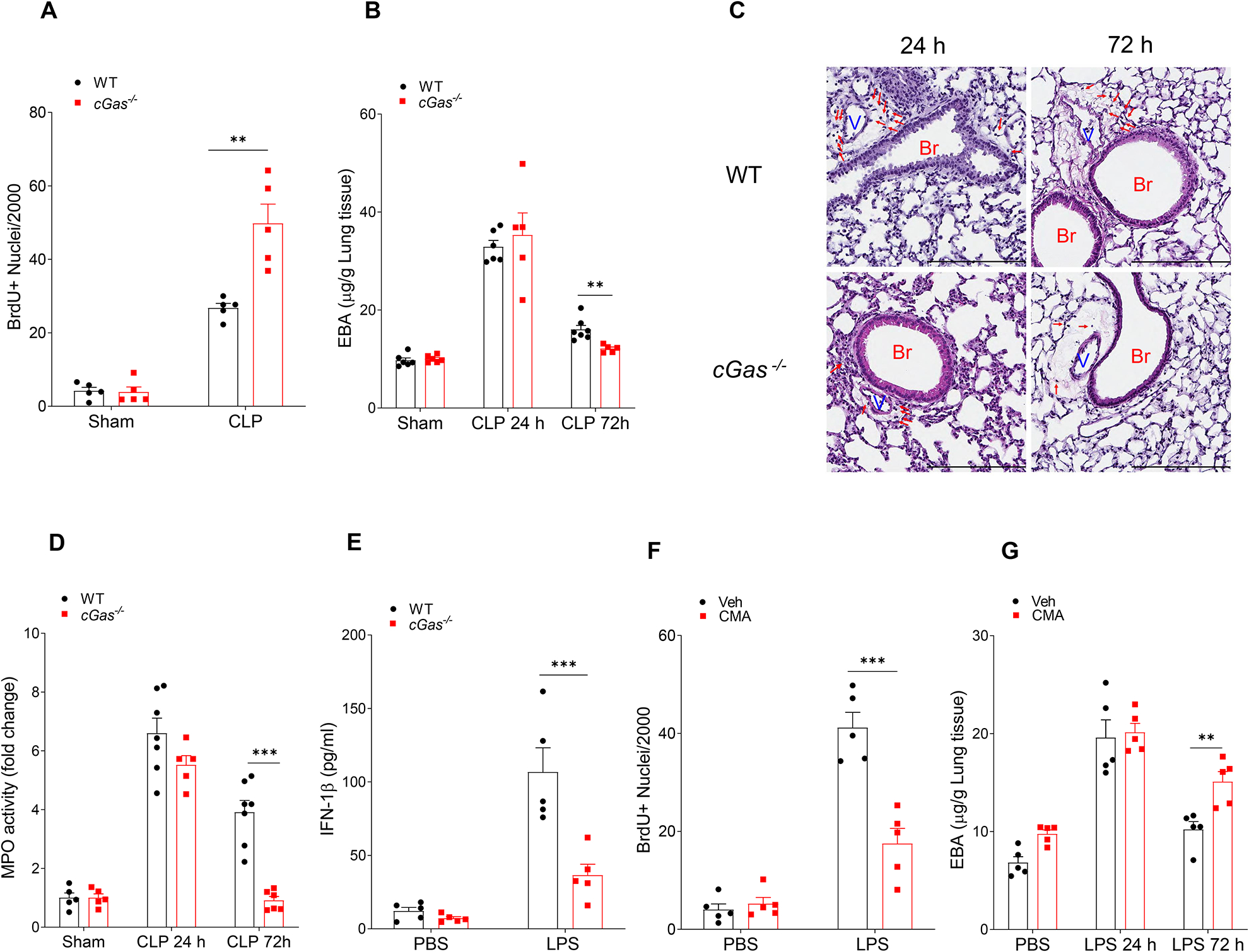
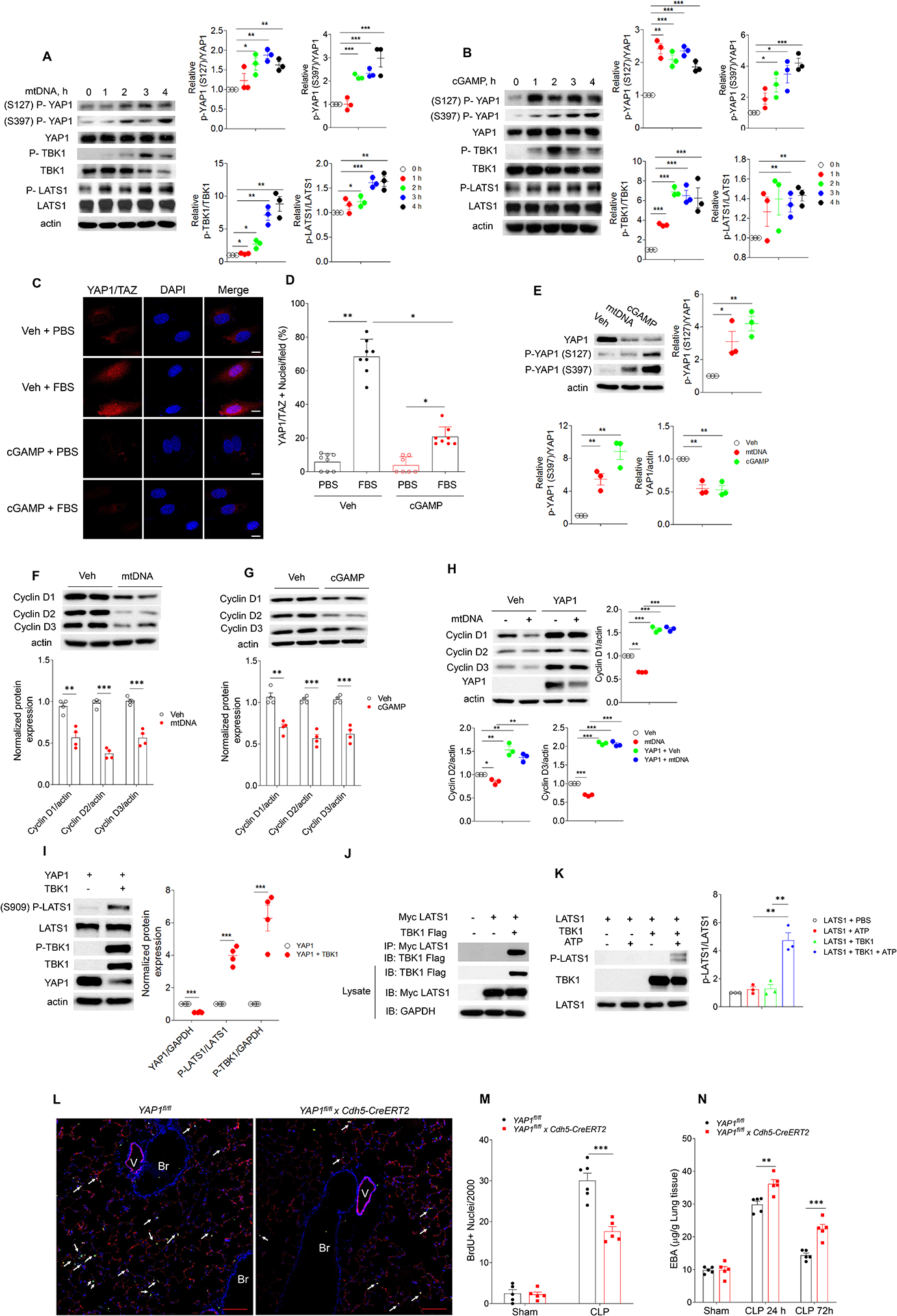
Cytosolic DNA acts as a universal danger-associated molecular pattern (DAMP) signal; however, the mechanisms of self-DNA release into the cytosol and its role in inflammatory tissue injury are not well understood. We found that the internalized bacterial endotoxin lipopolysaccharide (LPS) activated the pore-forming protein Gasdermin D, which formed mitochondrial pores and induced mitochondrial DNA (mtDNA) release into the cytosol of endothelial cells. mtDNA was recognized by the DNA sensor cGAS and generated the second messenger cGAMP, which suppressed endothelial cell proliferation by downregulating YAP1 signaling. This indicated that the surviving endothelial cells in the penumbrium of the inflammatory injury were compromised in their regenerative capacity. In an experimental model of inflammatory lung injury, deletion of cGas in mice restored endothelial regeneration. The results suggest that targeting the endothelial Gasdermin D activated cGAS-YAP signaling pathway could serve as a potential strategy for restoring endothelial function after inflammatory injury.
Keywords: Gasdermin D; YAP; cGAS; endothelial regeneration; inflammation; lung injury; mitochondrial DNA; pyroptosis; vascular injury.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration Of Interests The authors declare no competing interests.
Figures
Comment in
-
Mitochondrial GSDMD Pores DAMPen Pyroptosis.Immunity. 2020 Mar 17;52(3):424-426. doi: 10.1016/j.immuni.2020.02.012. Immunity. 2020. PMID: 32187511 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
