

Effect of helical kink in antimicrobial peptides on membrane pore formation
- PMID: 32167466
- PMCID: PMC7069690
- DOI: 10.7554/eLife.47946
Effect of helical kink in antimicrobial peptides on membrane pore formation
Abstract
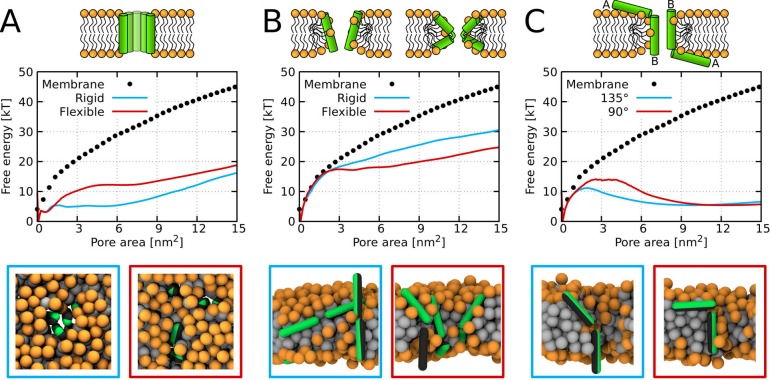
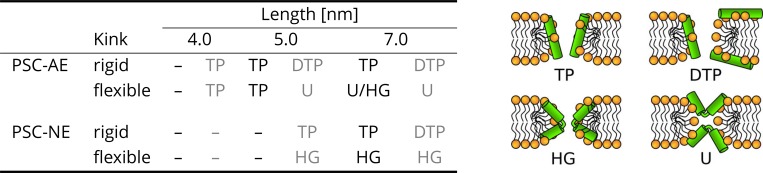
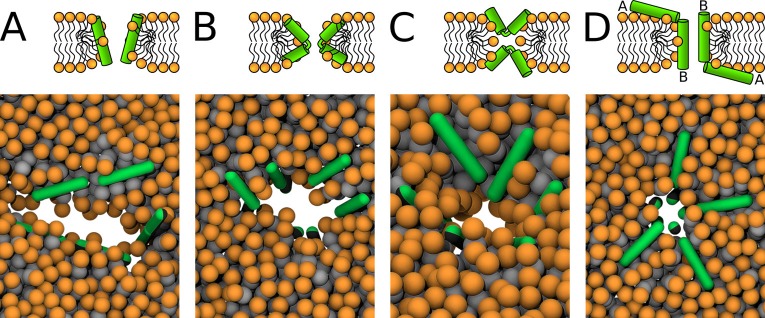
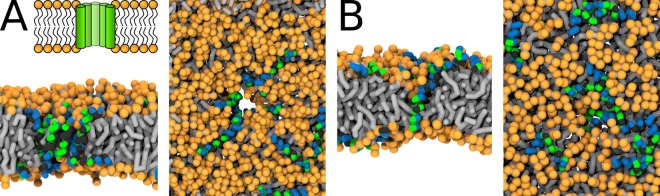
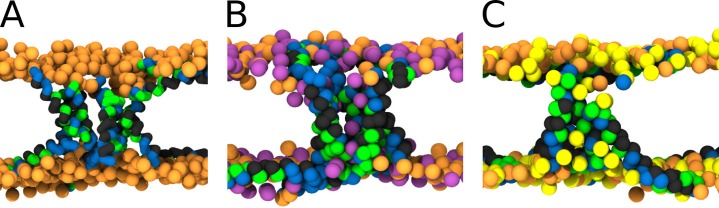
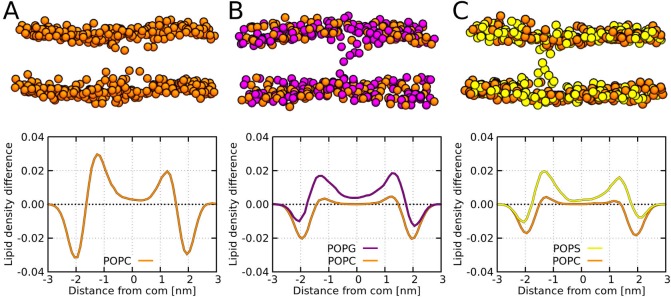
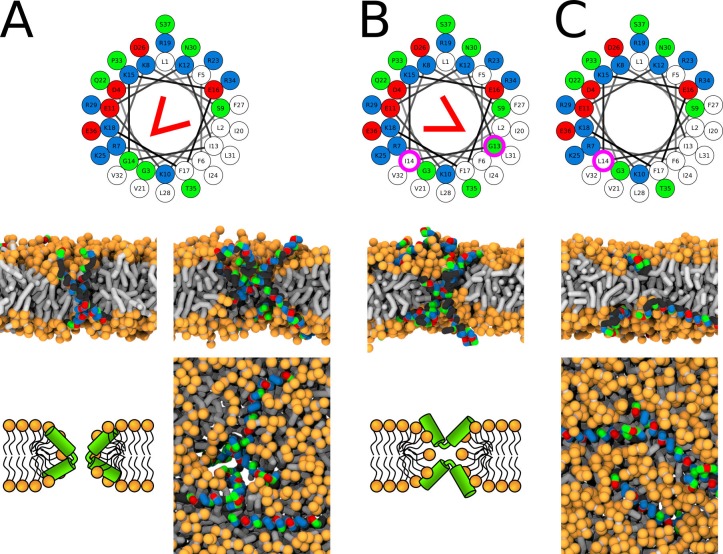
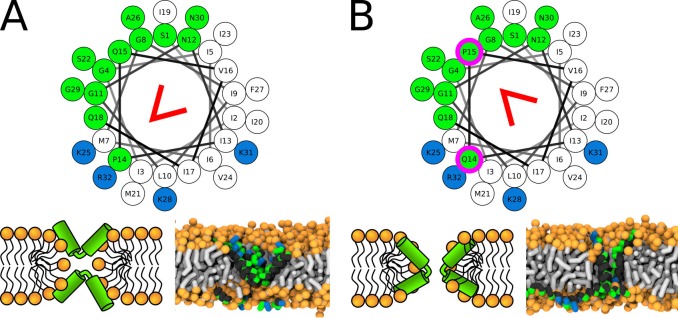
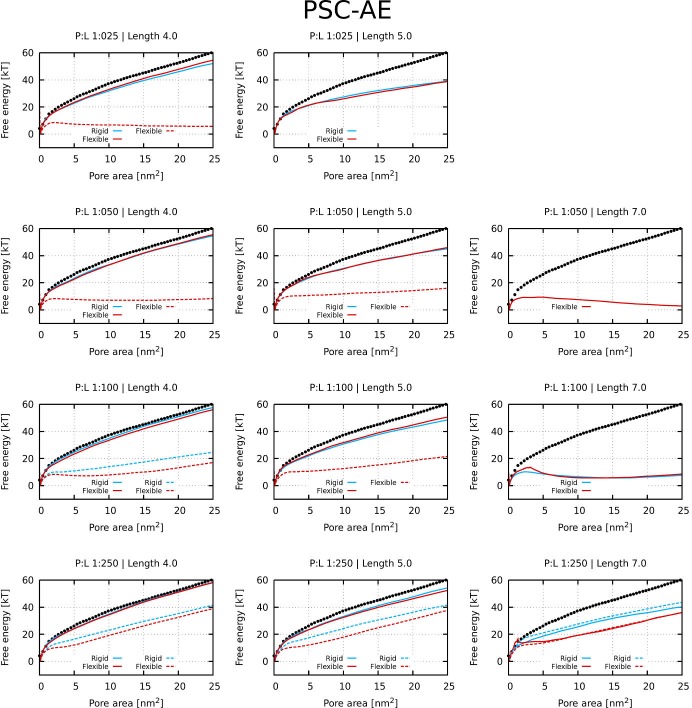
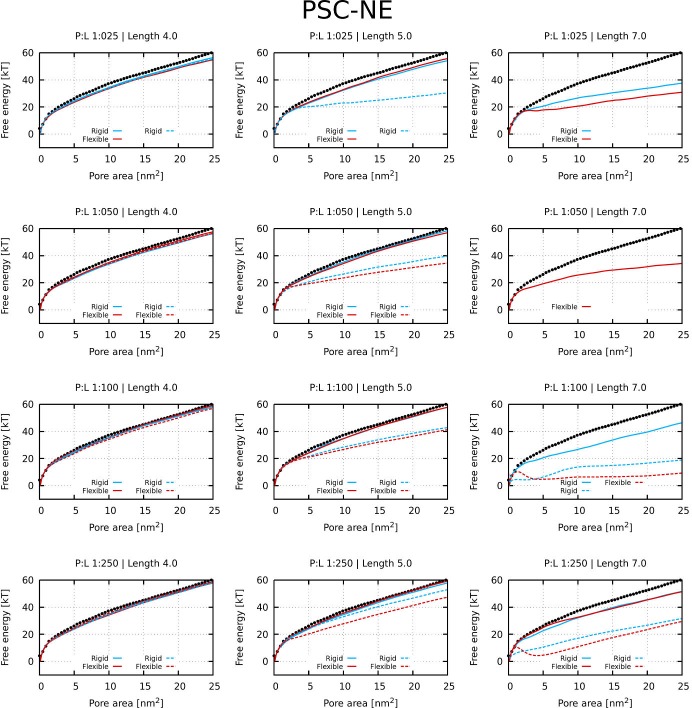
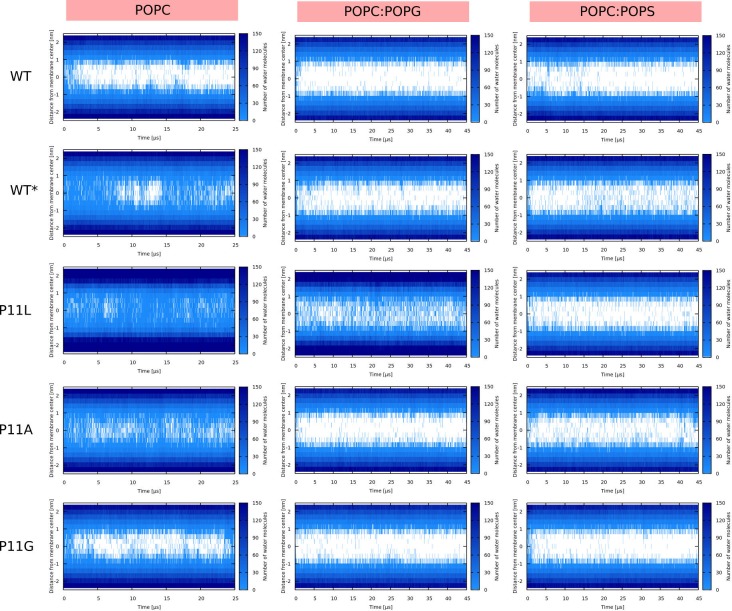
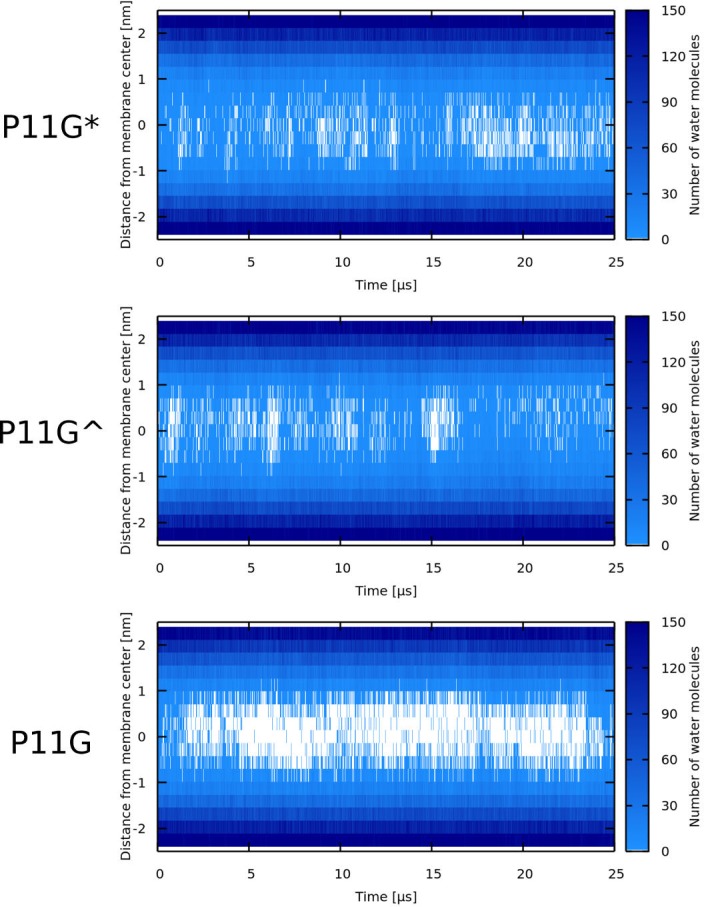
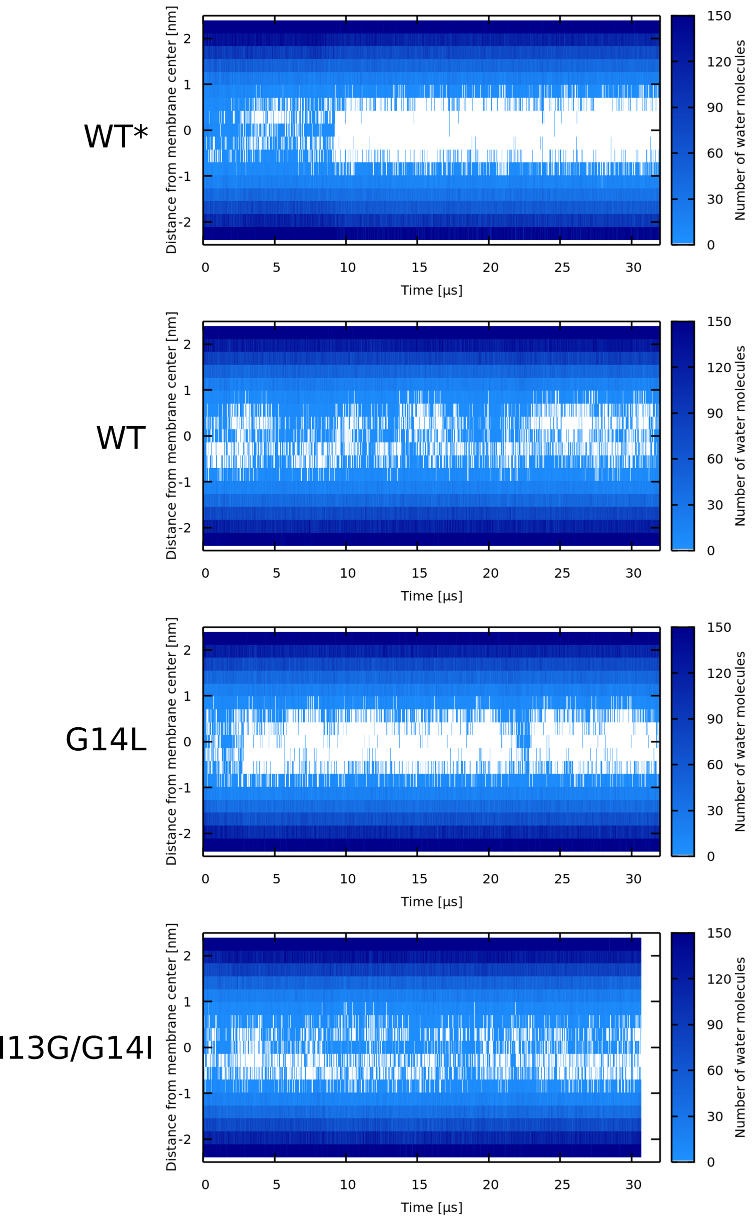
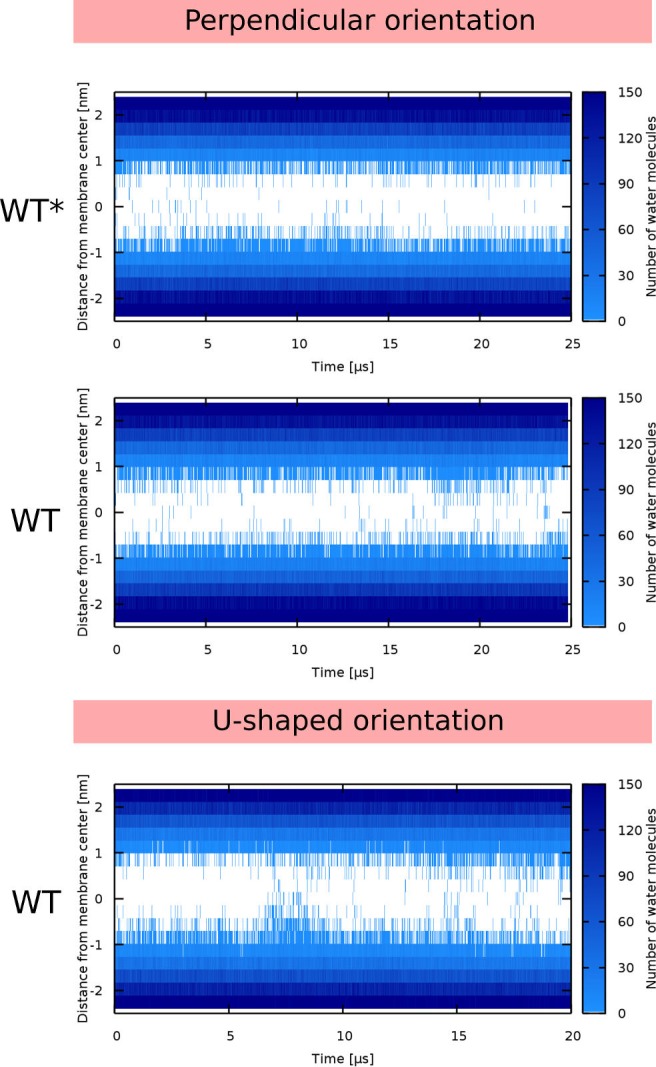
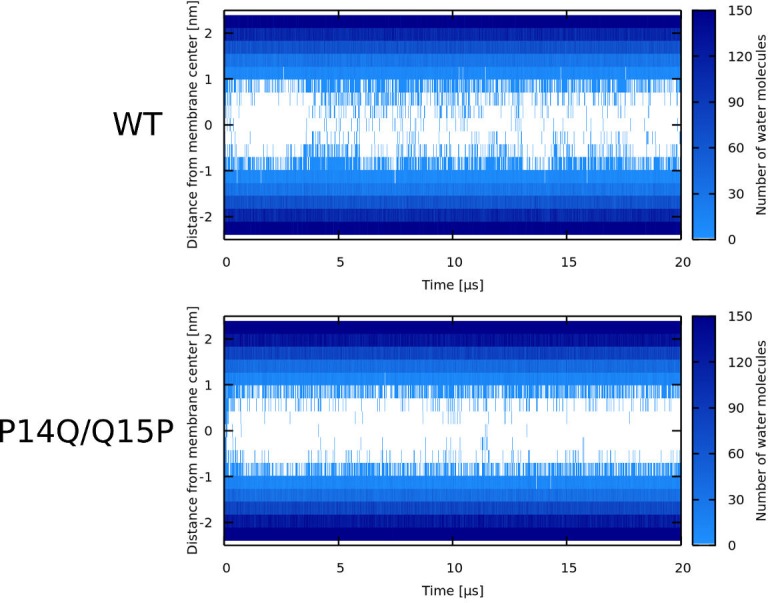
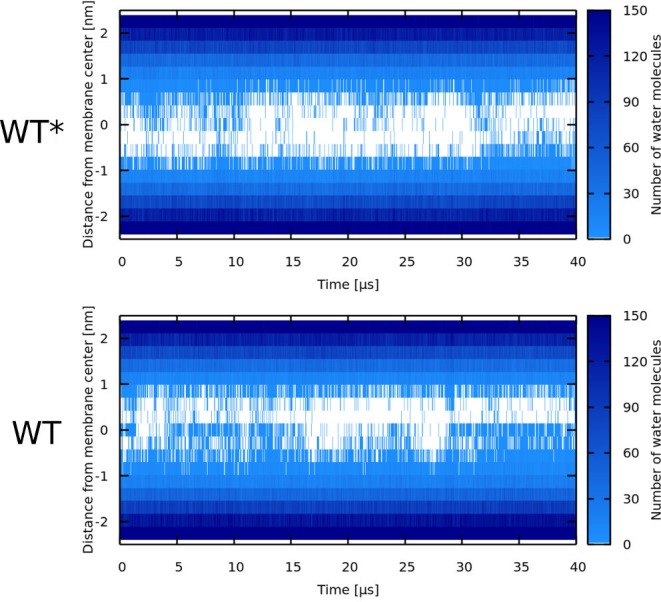
Every cell is protected by a semipermeable membrane. Peptides with the right properties, for example Antimicrobial peptides (AMPs), can disrupt this protective barrier by formation of leaky pores. Unfortunately, matching peptide properties with their ability to selectively form pores in bacterial membranes remains elusive. In particular, the proline/glycine kink in helical peptides was reported to both increase and decrease antimicrobial activity. We used computer simulations and fluorescence experiments to show that a kink in helices affects the formation of membrane pores by stabilizing toroidal pores but disrupting barrel-stave pores. The position of the proline/glycine kink in the sequence further controls the specific structure of toroidal pore. Moreover, we demonstrate that two helical peptides can form a kink-like connection with similar behavior as one long helical peptide with a kink. The provided molecular-level insight can be utilized for design and modification of pore-forming antibacterial peptides or toxins.
Keywords: antibiotics; fluorescent probes; membrane structure; membrane transport; molecular biophysics; none; structural biology.
© 2020, Tuerkova et al.
Conflict of interest statement
AT, IK, TK, LS, ŠP, MH, RV No competing interests declared
Figures
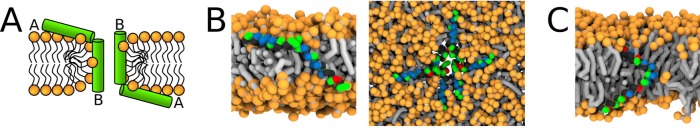
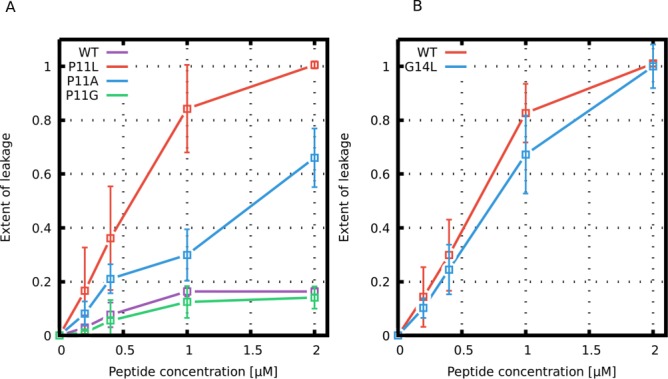
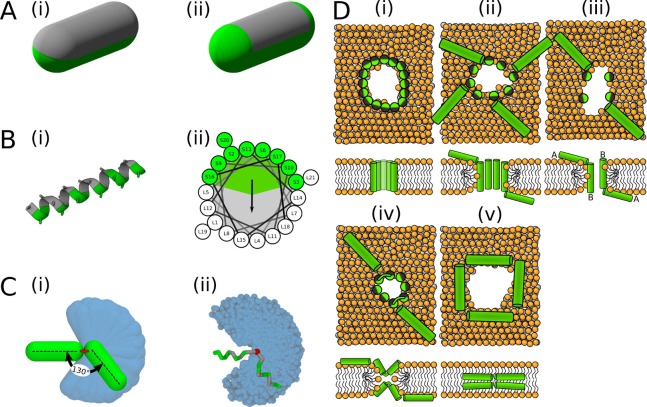
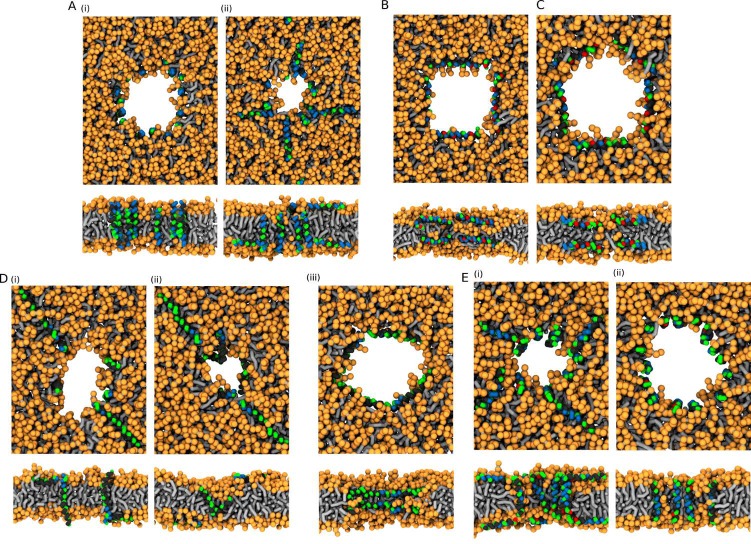
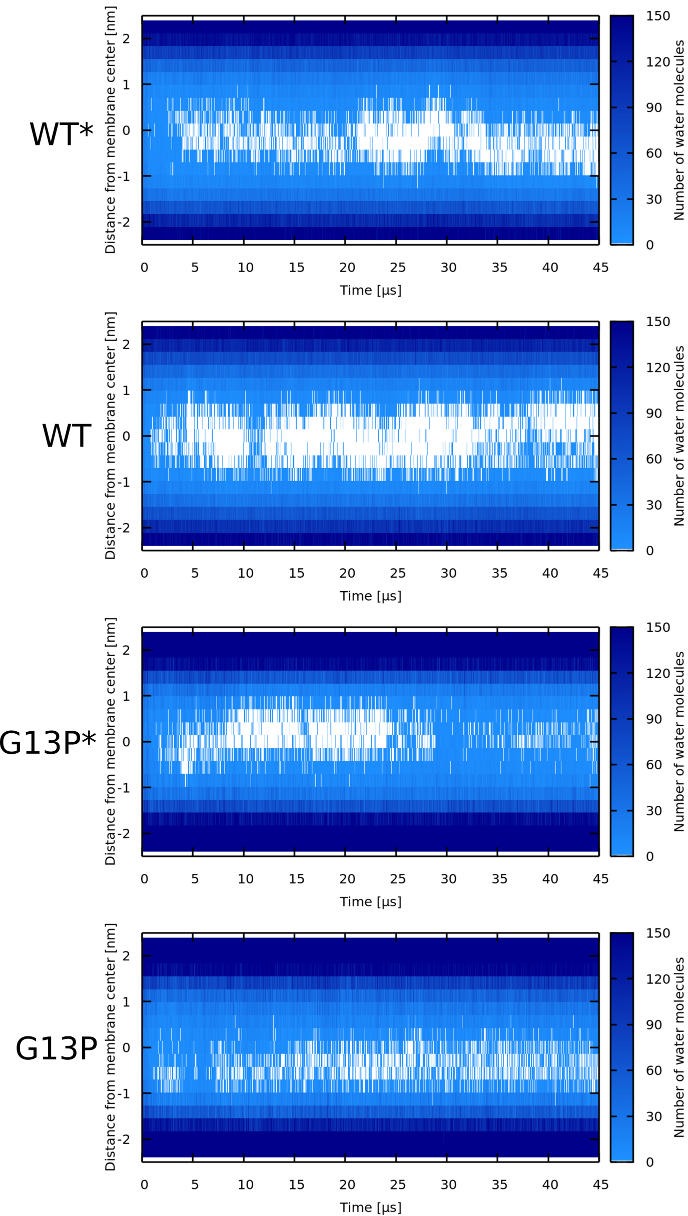
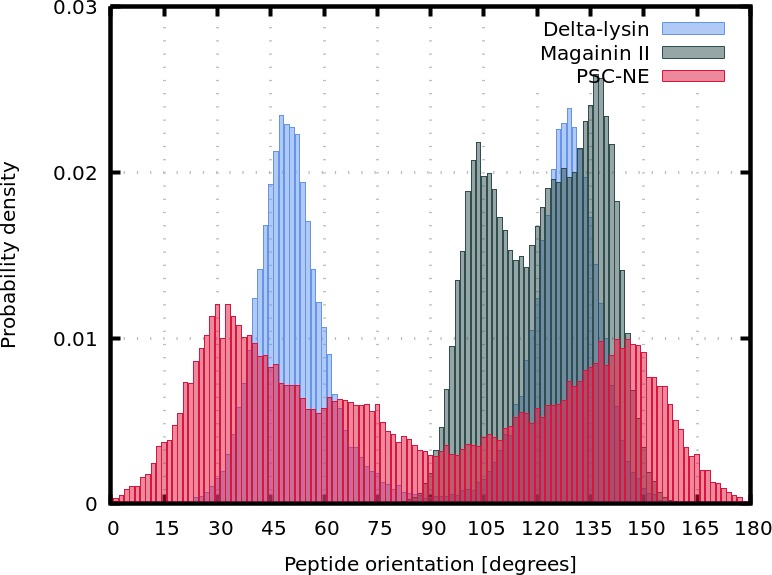
Similar articles
-
Advances in Molecular Understanding of α-Helical Membrane-Active Peptides.Acc Chem Res. 2021 May 4;54(9):2196-2204. doi: 10.1021/acs.accounts.1c00047. Epub 2021 Apr 12. Acc Chem Res. 2021. PMID: 33844916 Review.
-
Terminal charges modulate the pore forming activity of cationic amphipathic helices.Biochim Biophys Acta Biomembr. 2020 Apr 1;1862(4):183243. doi: 10.1016/j.bbamem.2020.183243. Epub 2020 Feb 29. Biochim Biophys Acta Biomembr. 2020. PMID: 32126225
-
The importance of membrane defects-lessons from simulations.Acc Chem Res. 2014 Aug 19;47(8):2244-51. doi: 10.1021/ar4002729. Epub 2014 Jun 3. Acc Chem Res. 2014. PMID: 24892900
-
Antimicrobial peptides in toroidal and cylindrical pores.Biochim Biophys Acta. 2010 Aug;1798(8):1485-93. doi: 10.1016/j.bbamem.2010.04.004. Epub 2010 Apr 18. Biochim Biophys Acta. 2010. PMID: 20403332 Free PMC article.
-
Antimicrobial peptides (AMPs): peptide structure and mode of action.J Biochem Mol Biol. 2005 Sep 30;38(5):507-16. doi: 10.5483/bmbrep.2005.38.5.507. J Biochem Mol Biol. 2005. Retraction in: J Biochem Mol Biol. 2005 Nov 30;38(6):766. PMID: 16202228 Retracted. Review.
Cited by
-
An optimized antimicrobial peptide analog acts as an antibiotic adjuvant to reverse methicillin-resistant Staphylococcus aureus.NPJ Sci Food. 2022 Dec 12;6(1):57. doi: 10.1038/s41538-022-00171-1. NPJ Sci Food. 2022. PMID: 36509755 Free PMC article.
-
Model architectures for bacterial membranes.Biophys Rev. 2022 Mar 7;14(1):111-143. doi: 10.1007/s12551-021-00913-7. eCollection 2022 Feb. Biophys Rev. 2022. PMID: 35340604 Free PMC article. Review.
-
Strategic Single-Residue Substitution in the Antimicrobial Peptide Esc(1-21) Confers Activity against Staphylococcus aureus, Including Drug-Resistant and Biofilm Phenotype.ACS Infect Dis. 2024 Jul 12;10(7):2403-2418. doi: 10.1021/acsinfecdis.4c00130. Epub 2024 Jun 7. ACS Infect Dis. 2024. PMID: 38848266 Free PMC article.
-
Photobuforin II, a fluorescent photoswitchable peptide.BBA Adv. 2023 Sep 29;4:100106. doi: 10.1016/j.bbadva.2023.100106. eCollection 2023. BBA Adv. 2023. PMID: 37842183 Free PMC article.
-
Novel Antimicrobial Peptide "Octoprohibitin" against Multidrug Resistant Acinetobacter baumannii.Pharmaceuticals (Basel). 2022 Jul 27;15(8):928. doi: 10.3390/ph15080928. Pharmaceuticals (Basel). 2022. PMID: 36015076 Free PMC article.
References
-
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1-2:19–25. doi: 10.1016/j.softx.2015.06.001. - DOI
-
- Alouf JE, Dufourcq J, Siffert O, Thiaudiere E, Geoffroy C. Interaction of staphylococcal delta-toxin and synthetic analogues with erythrocytes and phospholipid vesicles. biological and physical properties of the amphipathic peptides. European Journal of Biochemistry. 1989;183:381–390. doi: 10.1111/j.1432-1033.1989.tb14939.x. - DOI - PubMed
-
- Amos ST, Vermeer LS, Ferguson PM, Kozlowska J, Davy M, Bui TT, Drake AF, Lorenz CD, Mason AJ. Antimicrobial peptide potency is facilitated by greater conformational flexibility when binding to Gram-negative bacterial inner membranes. Scientific Reports. 2016;6:37639. doi: 10.1038/srep37639. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
