

GenomegaMap: Within-Species Genome-Wide dN/dS Estimation from over 10,000 Genomes
- PMID: 32167543
- PMCID: PMC7403622
- DOI: 10.1093/molbev/msaa069
GenomegaMap: Within-Species Genome-Wide dN/dS Estimation from over 10,000 Genomes
Abstract
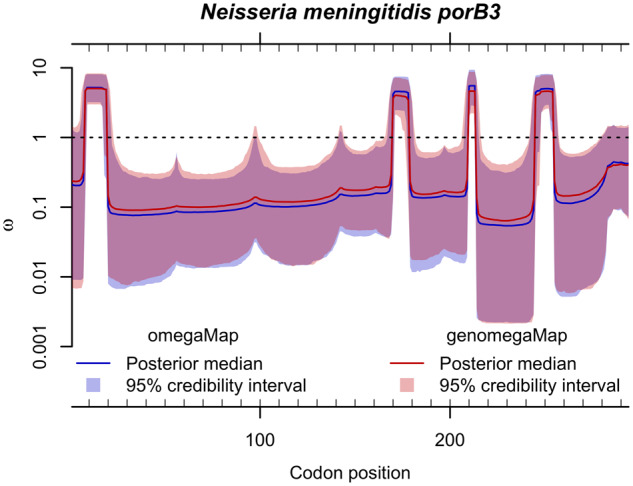
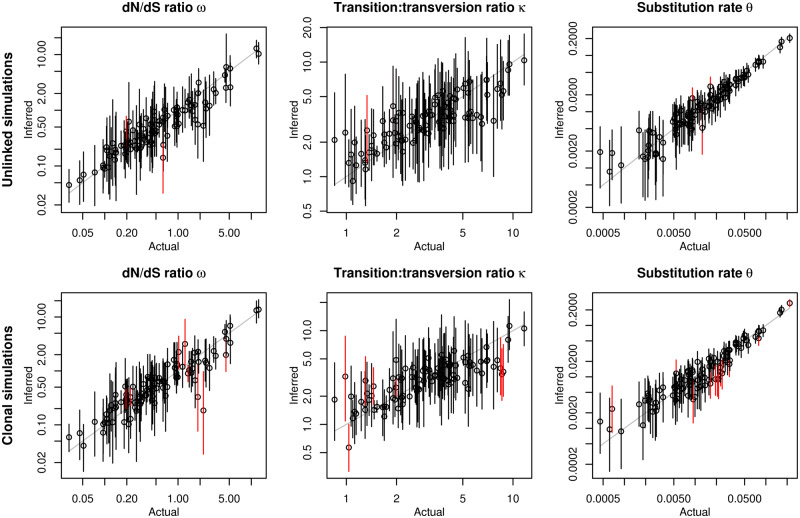
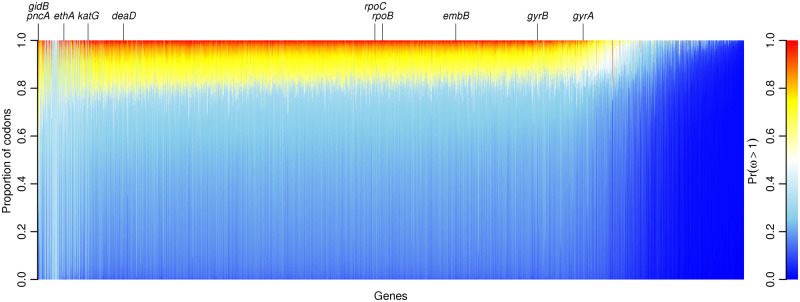
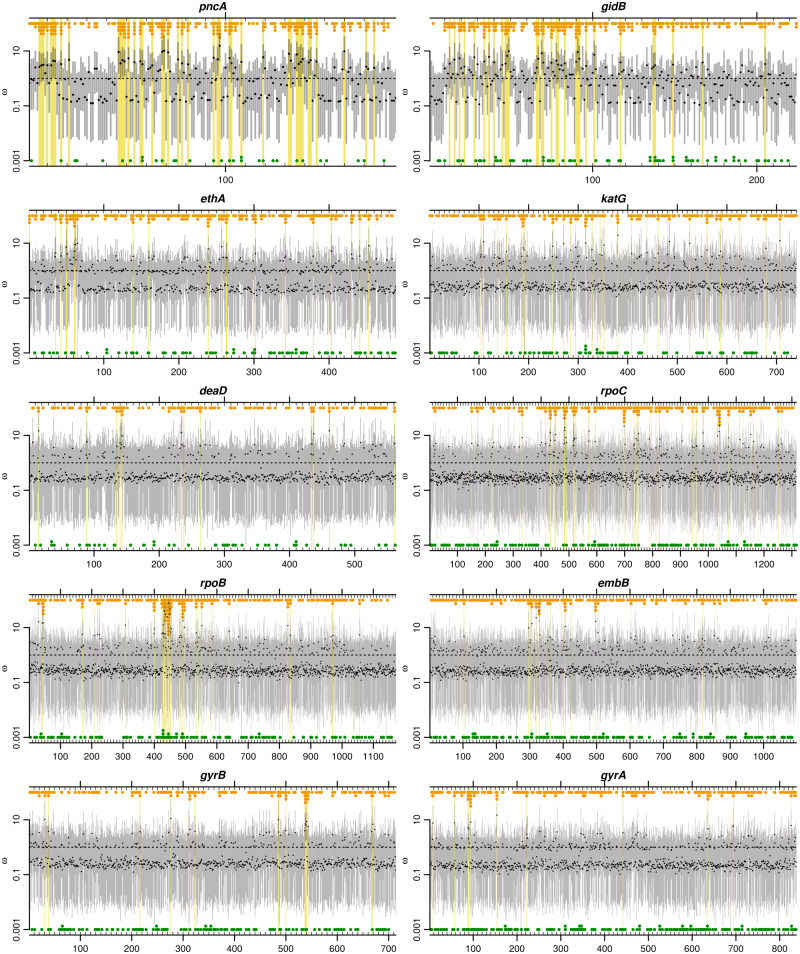
The dN/dS ratio provides evidence of adaptation or functional constraint in protein-coding genes by quantifying the relative excess or deficit of amino acid-replacing versus silent nucleotide variation. Inexpensive sequencing promises a better understanding of parameters, such as dN/dS, but analyzing very large data sets poses a major statistical challenge. Here, I introduce genomegaMap for estimating within-species genome-wide variation in dN/dS, and I apply it to 3,979 genes across 10,209 tuberculosis genomes to characterize the selection pressures shaping this global pathogen. GenomegaMap is a phylogeny-free method that addresses two major problems with existing approaches: 1) It is fast no matter how large the sample size and 2) it is robust to recombination, which causes phylogenetic methods to report artefactual signals of adaptation. GenomegaMap uses population genetics theory to approximate the distribution of allele frequencies under general, parent-dependent mutation models. Coalescent simulations show that substitution parameters are well estimated even when genomegaMap's simplifying assumption of independence among sites is violated. I demonstrate the ability of genomegaMap to detect genuine signatures of selection at antimicrobial resistance-conferring substitutions in Mycobacterium tuberculosis and describe a novel signature of selection in the cold-shock DEAD-box protein A gene deaD/csdA. The genomegaMap approach helps accelerate the exploitation of big data for gaining new insights into evolution within species.
Keywords: adaptation; big data; dN/dS; natural selection; parent-dependent mutation; recombination.
© The Author(s) 2020. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
-
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, et al. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393(6685):537–544., - PubMed
