

Uncovering sex-specific mechanisms of action of testosterone and redox balance
- PMID: 32169396
- PMCID: PMC7212492
- DOI: 10.1016/j.redox.2020.101490
Uncovering sex-specific mechanisms of action of testosterone and redox balance
Abstract
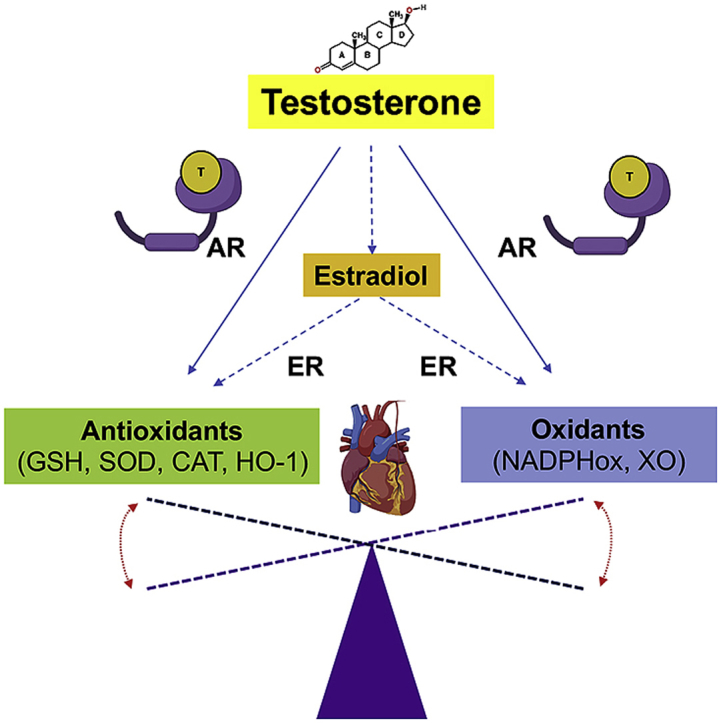
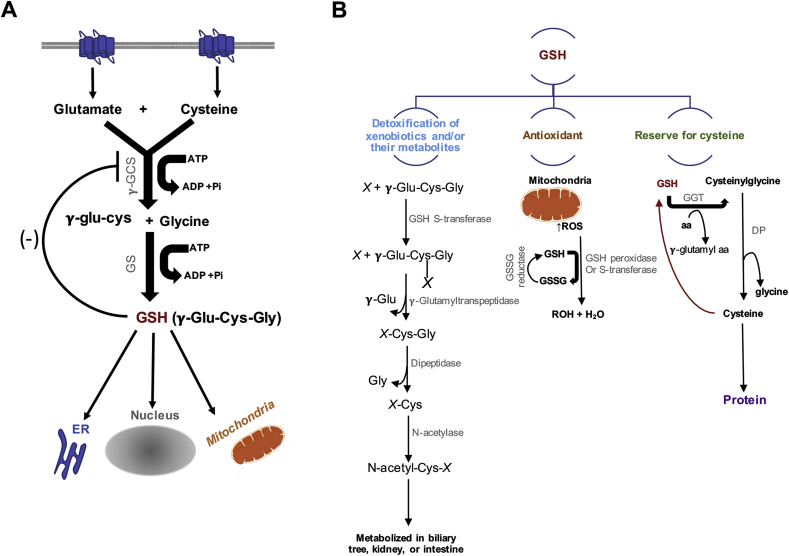
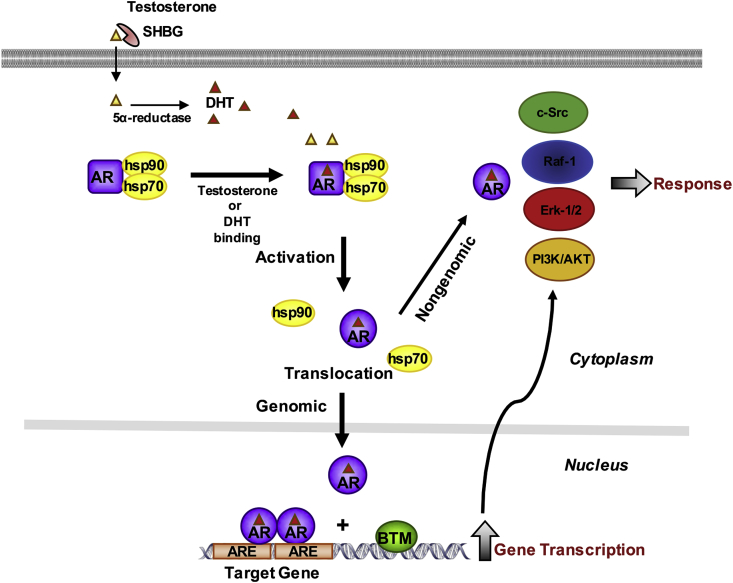
The molecular and pharmacological manipulation of the endogenous redox system is a promising therapy to limit myocardial damage after a heart attack; however, antioxidant therapies have failed to fully establish their cardioprotective effects, suggesting that additional factors, including antioxidant system interactions with other molecular pathways, may alter the pharmacological effects of antioxidants. Since gender differences in cardiovascular disease (CVD) are prevalent, and sex is an essential determinant of the response to oxidative stress, it is of particular interest to understand the effects of sex hormone signaling on the activity and expression of cellular antioxidants and the pharmacological actions of antioxidant therapies. In the present review, we briefly summarize the current understanding of testosterone effects on the modulation of the endogenous antioxidant systems in the CV system, cardiomyocytes, and the heart. We also review the latest research on redox balance and sexual dimorphism, with particular emphasis on the role of the natural antioxidant system glutathione (GSH) in the context of myocardial infarction, and the pro- and antioxidant effects of testosterone signaling via the androgen receptor (AR) on the heart. Finally, we discuss future perspectives regarding the potential of using combing antioxidant and testosterone replacement therapies to protect the aging myocardium.
Keywords: Androgen signaling; Cardiomyocytes; Glutathione; Heart; Oxidative stress; Testosterone.
Copyright © 2020 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of competing interest The author(s) declare(s) that there is no conflict of interest regarding the publication of this article.
Figures

References
-
- Benjamin E.J., Muntner P., Alonso A., Bittencourt M.S., Callaway C.W., Carson A.P., Chamberlain A.M., Chang A.R., Cheng S., Das S.R., Delling F.N., Djousse L., Elkind M.S.V., Ferguson J.F., Fornage M., Jordan L.C., Khan S.S., Kissela B.M., Knutson K.L., Kwan T.W., Lackland D.T., Lewis T.T., Lichtman J.H., Longenecker C.T., Loop M.S., Lutsey P.L., Martin S.S., Matsushita K., Moran A.E., Mussolino M.E., O'Flaherty M., Pandey A., Perak A.M., Rosamond W.D., Roth G.A., Sampson U.K.A., Satou G.M., Schroeder E.B., Shah S.H., Spartano N.L., Stokes A., Tirschwell D.L., Tsao C.W., Turakhia M.P., VanWagner L.B., Wilkins J.T., Wong S.S., Virani S.S. American heart association council on E, prevention statistics C and stroke statistics S. Heart disease and stroke statistics-2019 update: a report from the American heart association. Circulation. 2019;139:e56–e528. - PubMed
-
- Anderson R.D., Pepine C.J. Gender differences in the treatment for acute myocardial infarction: bias or biology? Circulation. 2007;115:823–826. - PubMed
-
- Diercks D.B., Owen K.P., Kontos M.C., Blomkalns A., Chen A.Y., Miller C., Wiviott S., Peterson E.D. Gender differences in time to presentation for myocardial infarction before and after a national women's cardiovascular awareness campaign: a temporal analysis from the can rapid risk stratification of unstable Angina patients suppress ADverse outcomes with early implementation (CRUSADE) and the national cardiovascular data registry acute coronary treatment and intervention outcomes network-get with the guidelines (NCDR ACTION registry-GWTG) Am. Heart J. 2010;160:80–87 e3. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
