

Ecosystem-wide metagenomic binning enables prediction of ecological niches from genomes
- PMID: 32170201
- PMCID: PMC7070063
- DOI: 10.1038/s42003-020-0856-x
Ecosystem-wide metagenomic binning enables prediction of ecological niches from genomes
Abstract
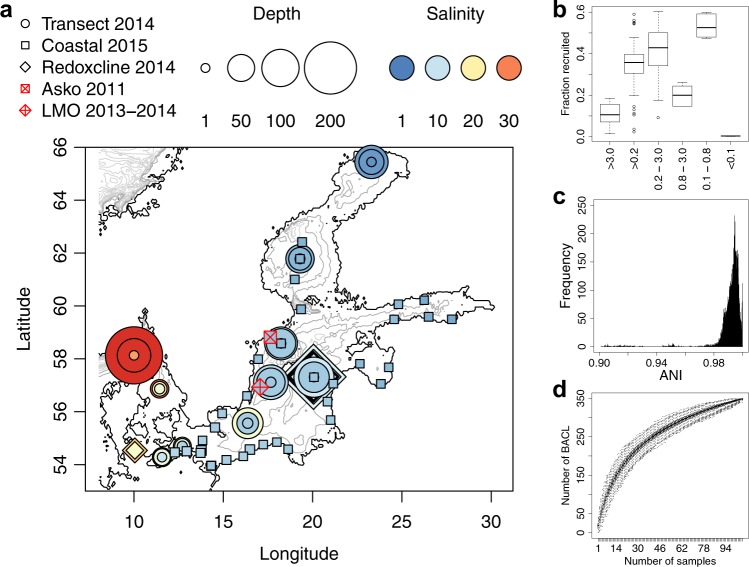
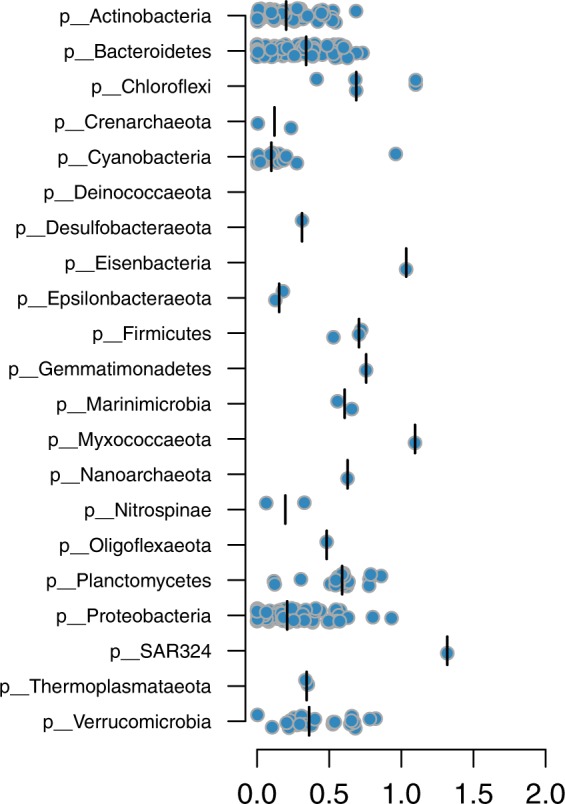
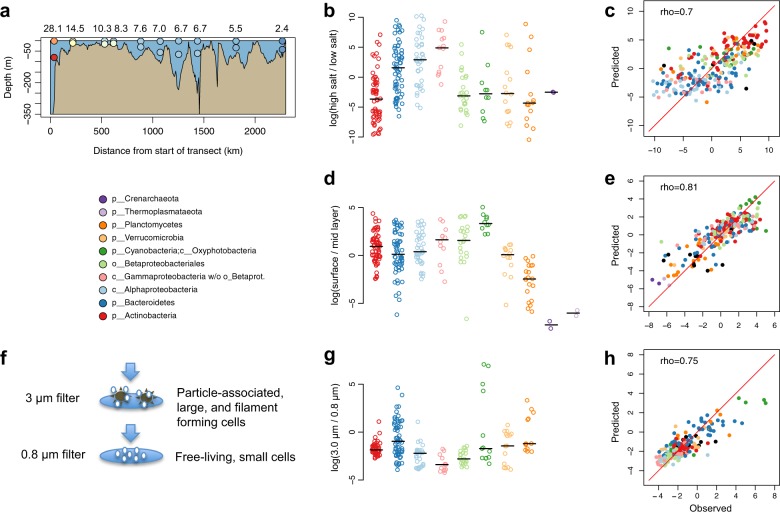
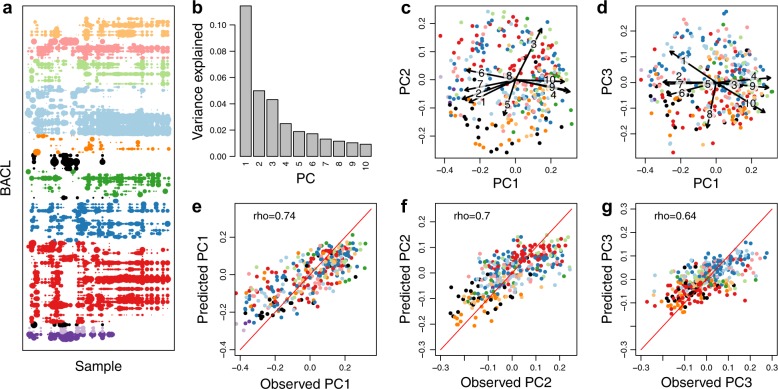
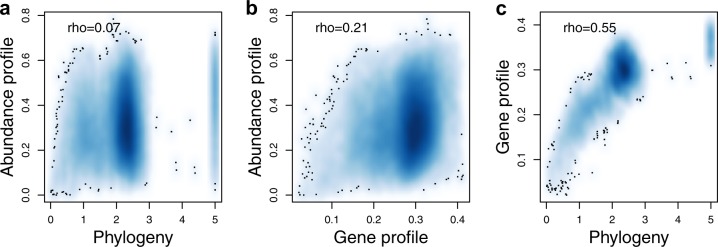
The genome encodes the metabolic and functional capabilities of an organism and should be a major determinant of its ecological niche. Yet, it is unknown if the niche can be predicted directly from the genome. Here, we conduct metagenomic binning on 123 water samples spanning major environmental gradients of the Baltic Sea. The resulting 1961 metagenome-assembled genomes represent 352 species-level clusters that correspond to 1/3 of the metagenome sequences of the prokaryotic size-fraction. By using machine-learning, the placement of a genome cluster along various niche gradients (salinity level, depth, size-fraction) could be predicted based solely on its functional genes. The same approach predicted the genomes' placement in a virtual niche-space that captures the highest variation in distribution patterns. The predictions generally outperformed those inferred from phylogenetic information. Our study demonstrates a strong link between genome and ecological niche and provides a conceptual framework for predictive ecology based on genomic data.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Hutchinson GE. Concluding remarks. Cold Spring Harb. Symposia Quant. Biol. 1957;22:415–427. doi: 10.1101/SQB.1957.022.01.039. - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
