

Hb S/ β-Thalassemia in the REDS-III Brazil Sickle Cell Disease Cohort: Clinical, Laboratory and Molecular Characteristics
- PMID: 32172616
- PMCID: PMC7225056
- DOI: 10.1080/03630269.2020.1731530
Hb S/ β-Thalassemia in the REDS-III Brazil Sickle Cell Disease Cohort: Clinical, Laboratory and Molecular Characteristics
Abstract
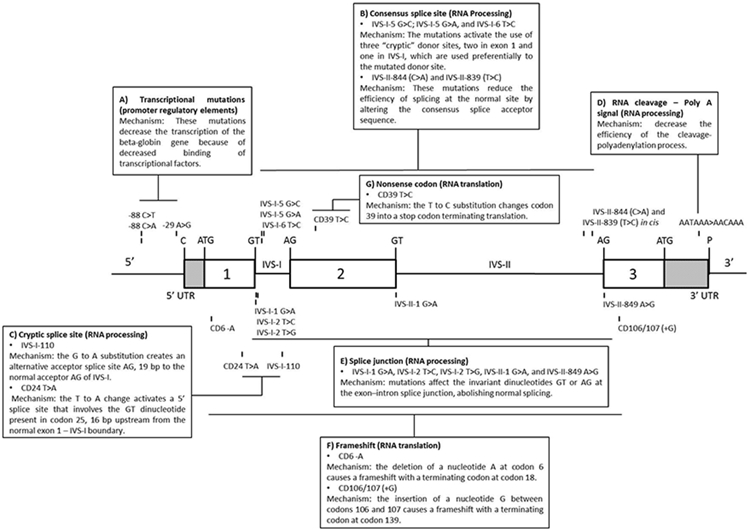
We described the clinical, laboratory and molecular characteristics of individuals with Hb S (HBB: c.20A>T)/β-thalassemia (Hb S/β-thal) participating in the Recipient Epidemiology and Donor Evaluation Study (REDS-III) Brazil Sickle Cell Disease cohort. HBB gene sequencing was performed to genotype each β-thal mutation. Patients were classified as Hb S/β0-thal, Hb S/β+-thal-severe or Hb S/β+-thal based on prior literature and databases of hemoglobin (Hb) variants. Characteristics of patients with each β-thal mutation were described and the clinical profile of patients grouped into Hb S/β0-thal, Hb S/β+-thal and Hb S/β+-thal-severe were compared. Of the 2793 patients enrolled, 84 (3.0%) had Hb S/β0-thal and 83 (3.0%) had Hb S/β+-thal; 40/83 (48.2%) patients with Hb S/β+-thal had mutations defined as severe. We identified 19 different β-thal mutations, eight Hb S/β0-thal, three Hb S/β+-thal-severe and eight Hb S/β+-thal. The most frequent β0 and β+ mutations were codon 39 (HBB: c.118C>T) and IVS-I-6 (T>C) (HBB: c.92+6T>C), respectively. Individuals with Hb S/β0-thal had a similar clinical and laboratory phenotype when compared to those with Hb S/β+-thal-severe. Individuals with Hb S/β+-thal-severe had significantly lower total Hb and Hb A levels and higher Hb S, white blood cell (WBC) count, platelets and hemolysis markers when compared to those with Hb S/β+-thal. Likewise, individuals with Hb S/β+-thal-severe showed a significantly higher occurrence of hospitalizations, vaso-occlusive events (VOE), acute chest syndrome (ACS), splenic sequestration, blood utilization, and hydroxyurea (HU) therapy.
Keywords: Clinical events; Hb S/β+-thalassemia (Hb S/β+-thal); Hb S/β0-thalassemia (Hb S/β0-thal); sickle cell disease; thalassemia mutation.
Conflict of interest statement
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Figures
References
-
- Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010;376(9757):2018–2031. - PubMed
-
- Serjeant GR, Sommereux AM, Stevenson M, et al. Comparison of sickle cell-β 0 thalassaemia with homozygous sickle cell disease. Br J Haematol. 1979;41(1):83–93. - PubMed
-
- Serjeant GR, Serjeant BE, Fraser RA, et al. Hb S-β-thalassemia: molecular, hematological and clinical comparisons. Hemoglobin. 2011;35(1):1–12. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical