

Identification of Two Novel Mutations in COG5 Causing Congenital Disorder of Glycosylation
- PMID: 32174980
- PMCID: PMC7056739
- DOI: 10.3389/fgene.2020.00168
Identification of Two Novel Mutations in COG5 Causing Congenital Disorder of Glycosylation
Abstract
Objective: This study reports a Chinese patient with a Congenital Disorder of Glycosylation (CDG) caused by compound-heterozygous mutations in the Conserved Oligomeric Golgi 5 (COG5) gene and thereby offers concrete evidence for early diagnosis.
Methods: The clinical manifestations, the results of laboratory examinations and genetic analysis of a 4-year-old Chinese girl with CDG are reported. We also reviewed previous CDG cases that involved COG5 mutations by comparing the phenotypes and genotypes in different cases.
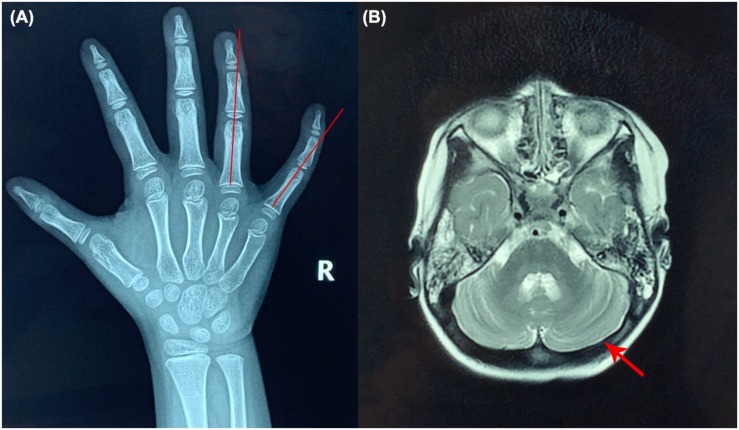
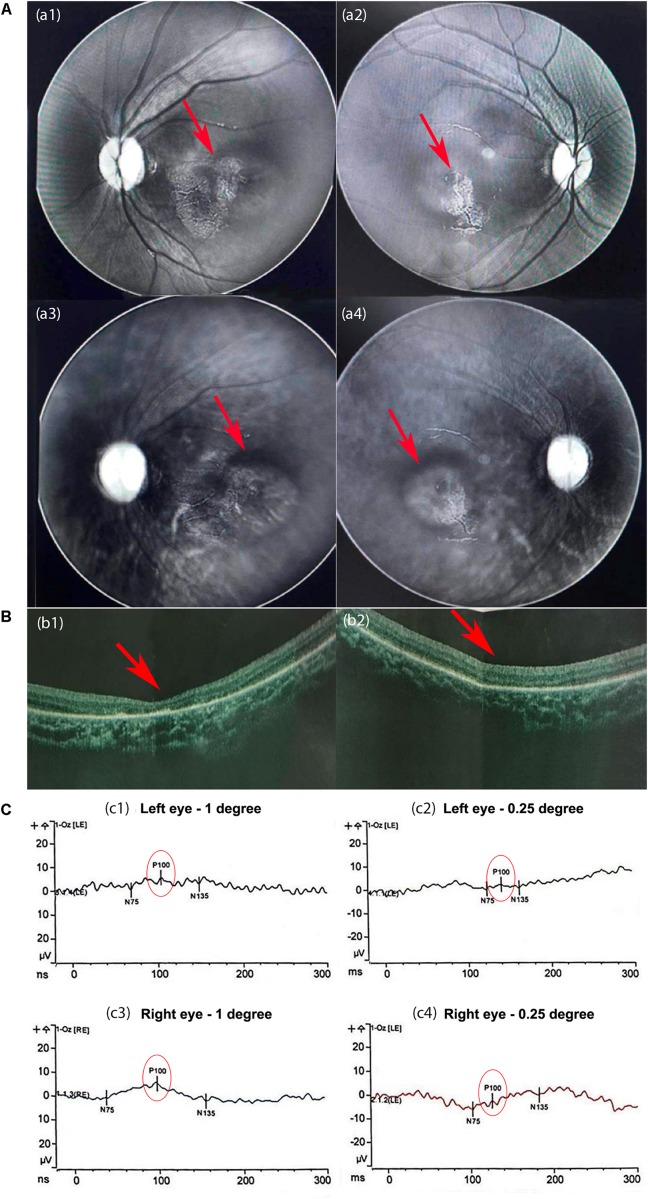
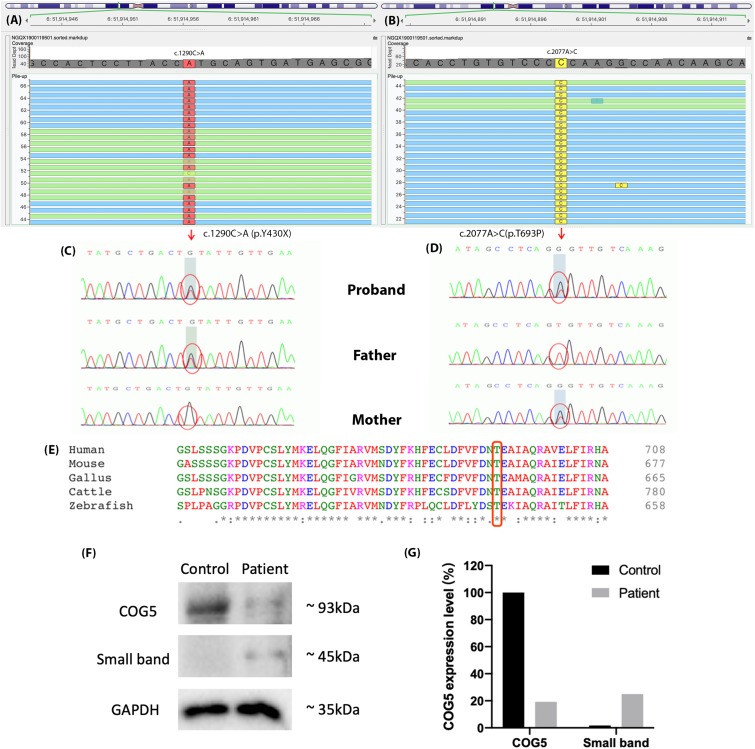
Results: The patient was admitted to our hospital due to ataxia and psychomotor delay. The major clinical manifestations were postural instability, difficulty in walking, psychomotor delay, hypohidrosis, hyperkeratosis of the skin, and ulnar deviation of the right-hand fingers. Biochemical analyses revealed coagulation defect and liver lesions. Vision tests showed choroidopathy and macular hypoplasia. Whole-exome sequencing identified the hitherto unreported compound-heterozygous COG5 mutations, c.1290C > A (p.Y430X) and c.2077A > C (p.T693P). Mutation p.Y430X is nonsense, leading to a truncated protein. Mutation p.T693P is located at a highly conserved region, and thus the polar-to-non-polar substitution presumably affects the structure and function of COG5. According to the Human Genome Mutation Database Professional, there have been totally 13 CDG cases caused by 13 COG5 mutations. They are mainly characterized by psychomotor delay, hypotonia, ataxia, microcephaly, and hearing and visual abnormalities.
Conclusion: The clinical manifestations of the patient are mild but consistent with the clinical characteristics of the published COG5-CDG cases. The results of this study extend the spectrum of clinical and genetic findings in COG5-CDG.
Keywords: COG5 gene; ataxia; congenital disorder of glycosylation; genetic sequencing; psychomotor delay; visual abnormalities.
Copyright © 2020 Wang, Han, Wang, Wang, Li, Jin and Wang.
Figures
Similar articles
-
Novel mutation of COG5 in a Taiwanese girl with congenital disorders of glycosylation manifesting as developmental delay.Mol Genet Metab Rep. 2024 Mar 22;39:101072. doi: 10.1016/j.ymgmr.2024.101072. eCollection 2024 Jun. Mol Genet Metab Rep. 2024. PMID: 38559322 Free PMC article.
-
Genetic analysis and prenatal diagnosis in a Chinese with growth retardation, abnormal liver function, and microcephaly.Mol Genet Genomic Med. 2021 Sep;9(9):e1751. doi: 10.1002/mgg3.1751. Epub 2021 Jul 31. Mol Genet Genomic Med. 2021. PMID: 34331832 Free PMC article. Review.
-
COG5-CDG: expanding the clinical spectrum.Orphanet J Rare Dis. 2012 Dec 10;7:94. doi: 10.1186/1750-1172-7-94. Orphanet J Rare Dis. 2012. PMID: 23228021 Free PMC article.
-
Novel compound heterozygous COG5 mutations in a Chinese male patient with severe clinical symptoms and type IIi congenital disorder of glycosylation: A case report.Exp Ther Med. 2019 Oct;18(4):2695-2700. doi: 10.3892/etm.2019.7834. Epub 2019 Jul 30. Exp Ther Med. 2019. PMID: 31572517 Free PMC article.
-
A novel variant in ALG1 gene associated with congenital disorder of glycosylation: A case report and short literature review.Mol Genet Genomic Med. 2023 Aug;11(8):e2197. doi: 10.1002/mgg3.2197. Epub 2023 May 19. Mol Genet Genomic Med. 2023. PMID: 37204045 Free PMC article. Review.
Cited by
-
Novel mutation of COG5 in a Taiwanese girl with congenital disorders of glycosylation manifesting as developmental delay.Mol Genet Metab Rep. 2024 Mar 22;39:101072. doi: 10.1016/j.ymgmr.2024.101072. eCollection 2024 Jun. Mol Genet Metab Rep. 2024. PMID: 38559322 Free PMC article.
-
Neonatal presentation of COG6-CDG with prominent skin phenotype.JIMD Rep. 2020 Aug 7;55(1):51-58. doi: 10.1002/jmd2.12154. eCollection 2020 Sep. JIMD Rep. 2020. PMID: 32905044 Free PMC article.
-
Genetic analysis and prenatal diagnosis in a Chinese with growth retardation, abnormal liver function, and microcephaly.Mol Genet Genomic Med. 2021 Sep;9(9):e1751. doi: 10.1002/mgg3.1751. Epub 2021 Jul 31. Mol Genet Genomic Med. 2021. PMID: 34331832 Free PMC article. Review.
-
Screening of the TMEM151A Gene in Patients With Paroxysmal Kinesigenic Dyskinesia and Other Movement Disorders.Front Neurol. 2022 May 30;13:865690. doi: 10.3389/fneur.2022.865690. eCollection 2022. Front Neurol. 2022. PMID: 35707035 Free PMC article.
-
Characterization of a missense variant in COG5 in a Tunisian patient with COG5-CDG syndrome and insights into the effect of non-synonymous variants on COG5 protein.J Hum Genet. 2024 Nov;69(11):591-597. doi: 10.1038/s10038-024-01273-2. Epub 2024 Jul 11. J Hum Genet. 2024. PMID: 38987656
References
-
- Altassan R., Peanne R., Jaeken J., Barone R., Bidet M., Borgel D., et al. (2019). International clinical guidelines for the management of phosphomannomutase 2-congenital disorders of glycosylation: diagnosis, treatment and follow up. J. Inherit. Metab. Dis. 42 5–28. - PubMed
-
- Conoley J., Impera J. E. (1995). “Gesell child developmental age scale,” in The Twelfth Mental Measurements Yearbook, eds Conoley J. C., Impara J. C. (Nebraska: Buros Institute; ).
LinkOut - more resources
Full Text Sources
Molecular Biology Databases