

Reduction of Substrates by Nitrogenases
- PMID: 32176472
- PMCID: PMC7703680
- DOI: 10.1021/acs.chemrev.9b00556
Reduction of Substrates by Nitrogenases
Abstract
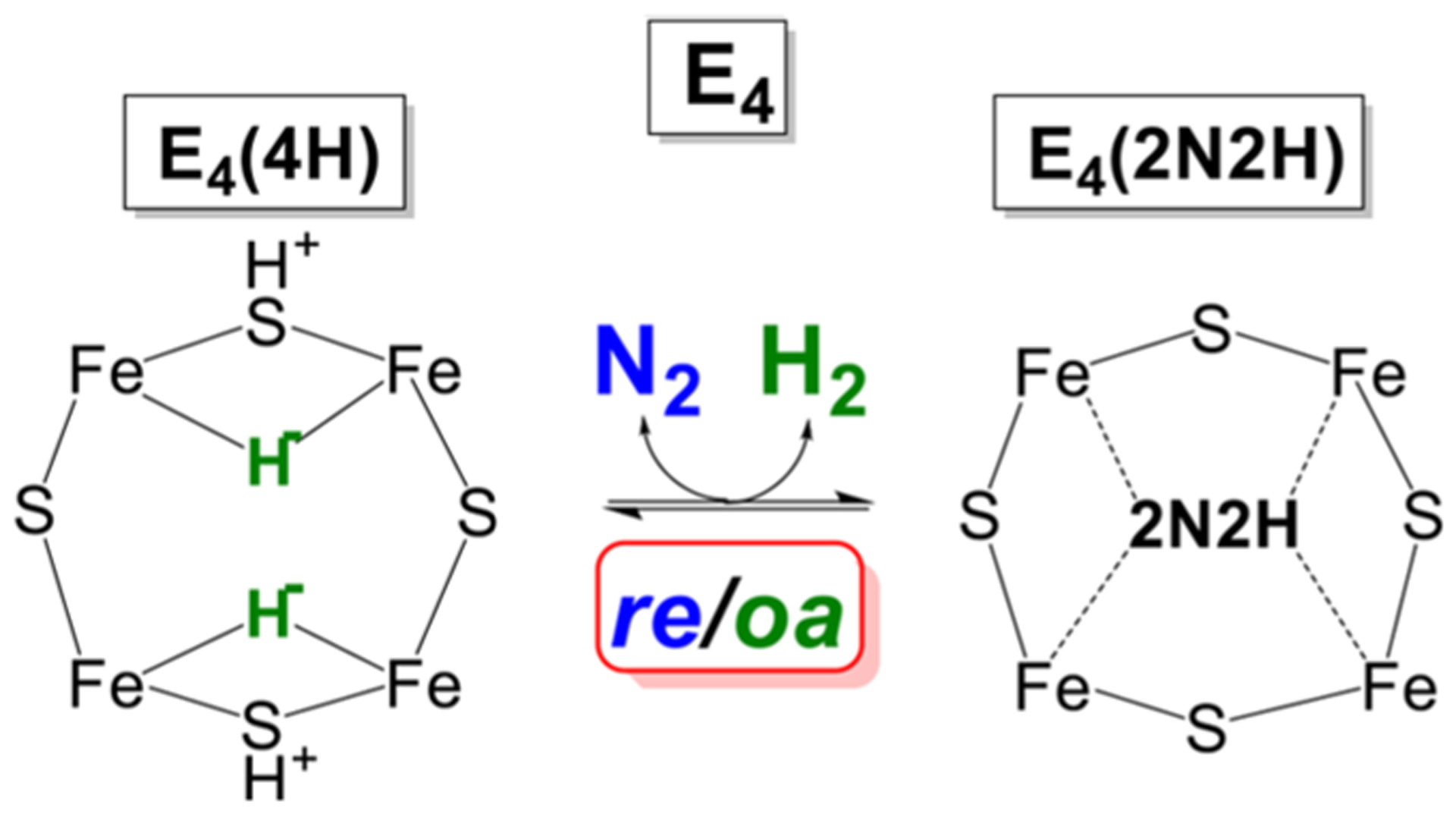
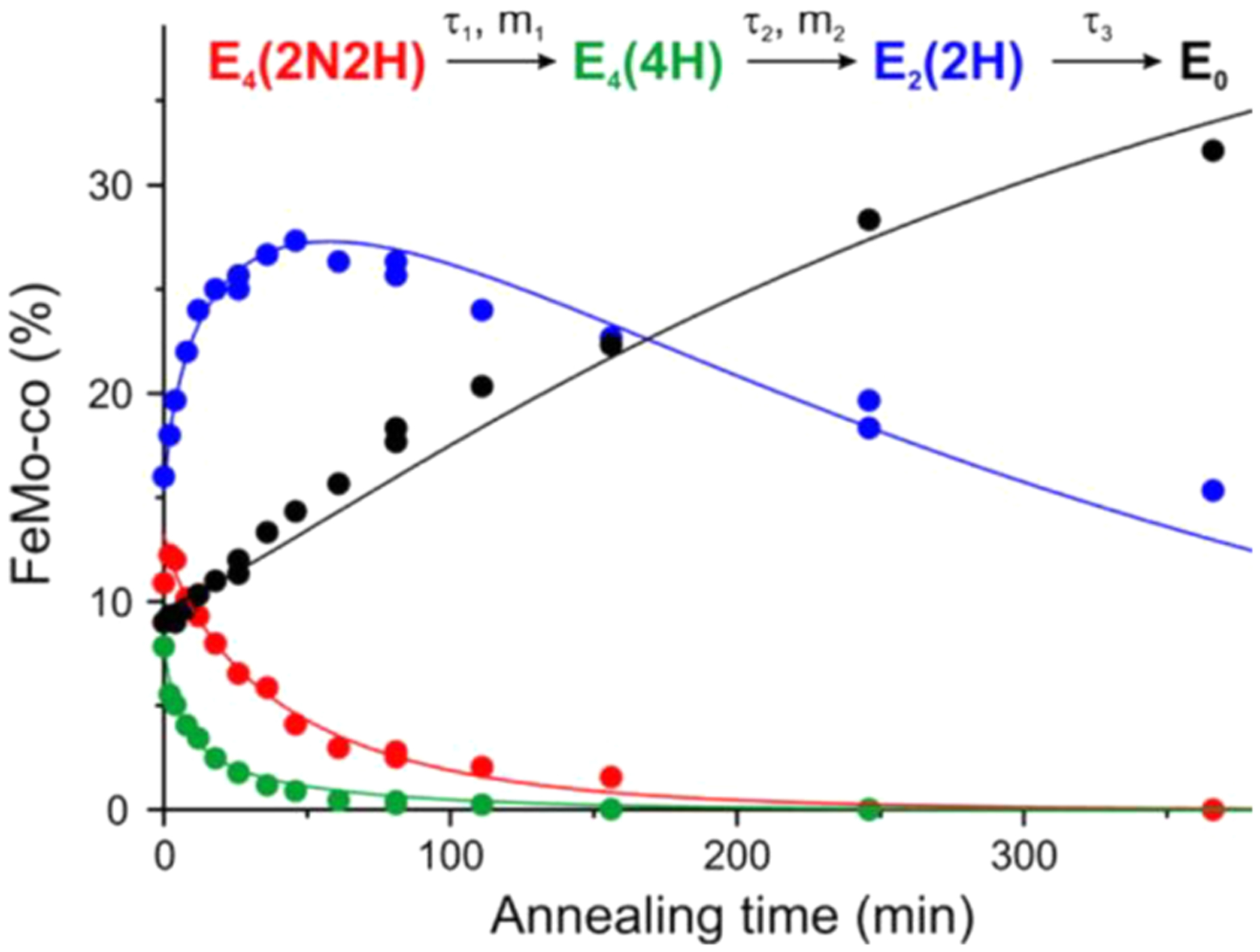
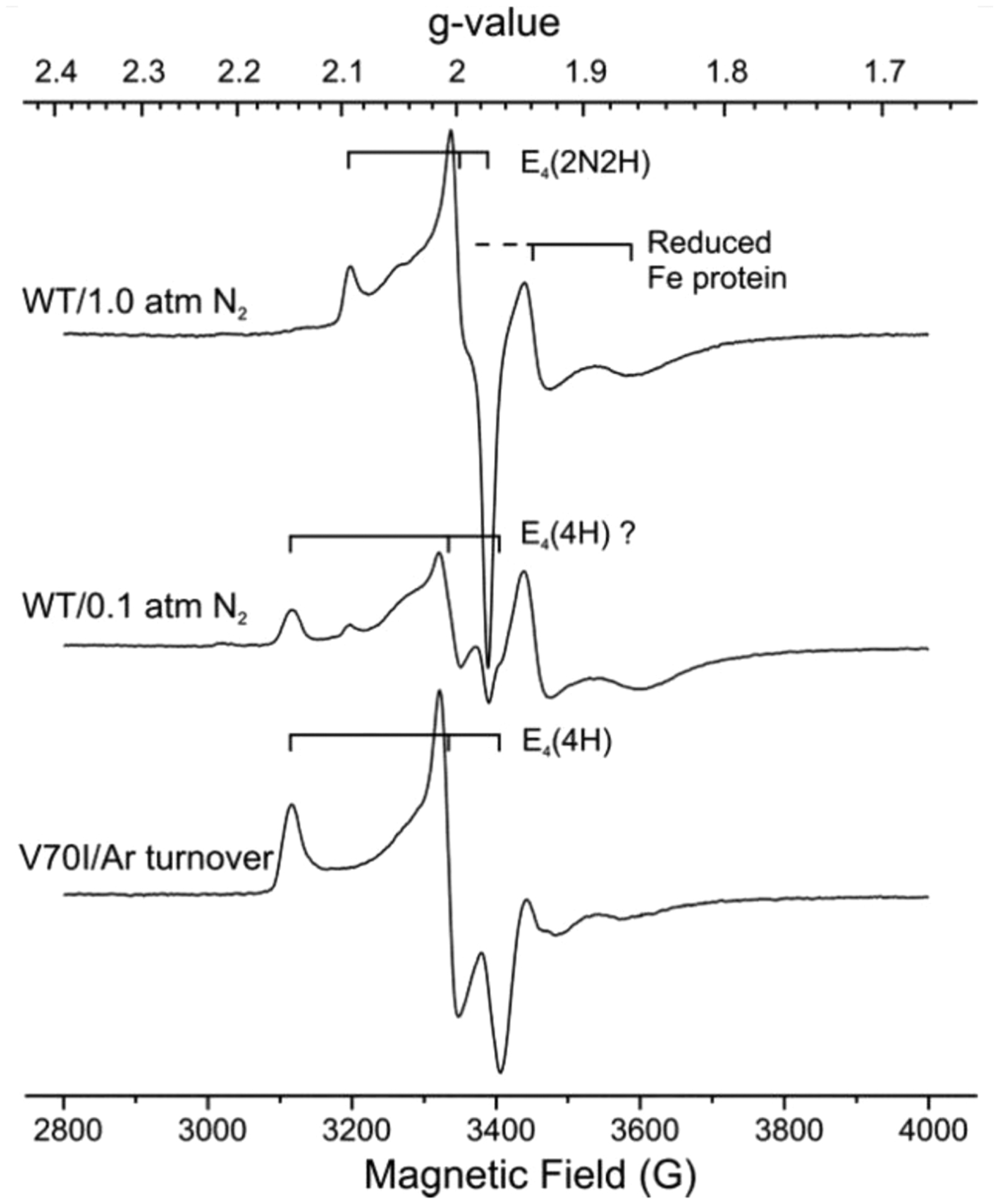
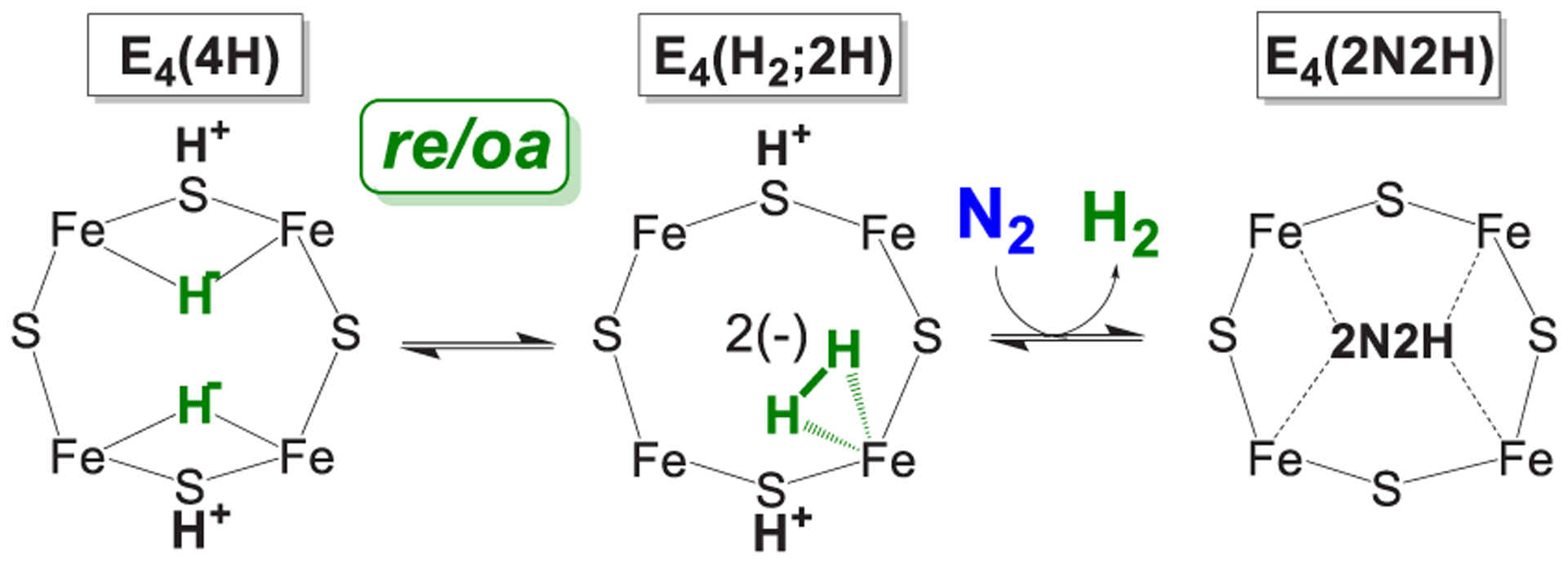
Nitrogenase is the enzyme that catalyzes biological N2 reduction to NH3. This enzyme achieves an impressive rate enhancement over the uncatalyzed reaction. Given the high demand for N2 fixation to support food and chemical production and the heavy reliance of the industrial Haber-Bosch nitrogen fixation reaction on fossil fuels, there is a strong need to elucidate how nitrogenase achieves this difficult reaction under benign conditions as a means of informing the design of next generation synthetic catalysts. This Review summarizes recent progress in addressing how nitrogenase catalyzes the reduction of an array of substrates. New insights into the mechanism of N2 and proton reduction are first considered. This is followed by a summary of recent gains in understanding the reduction of a number of other nitrogenous compounds not considered to be physiological substrates. Progress in understanding the reduction of a wide range of C-based substrates, including CO and CO2, is also discussed, and remaining challenges in understanding nitrogenase substrate reduction are considered.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
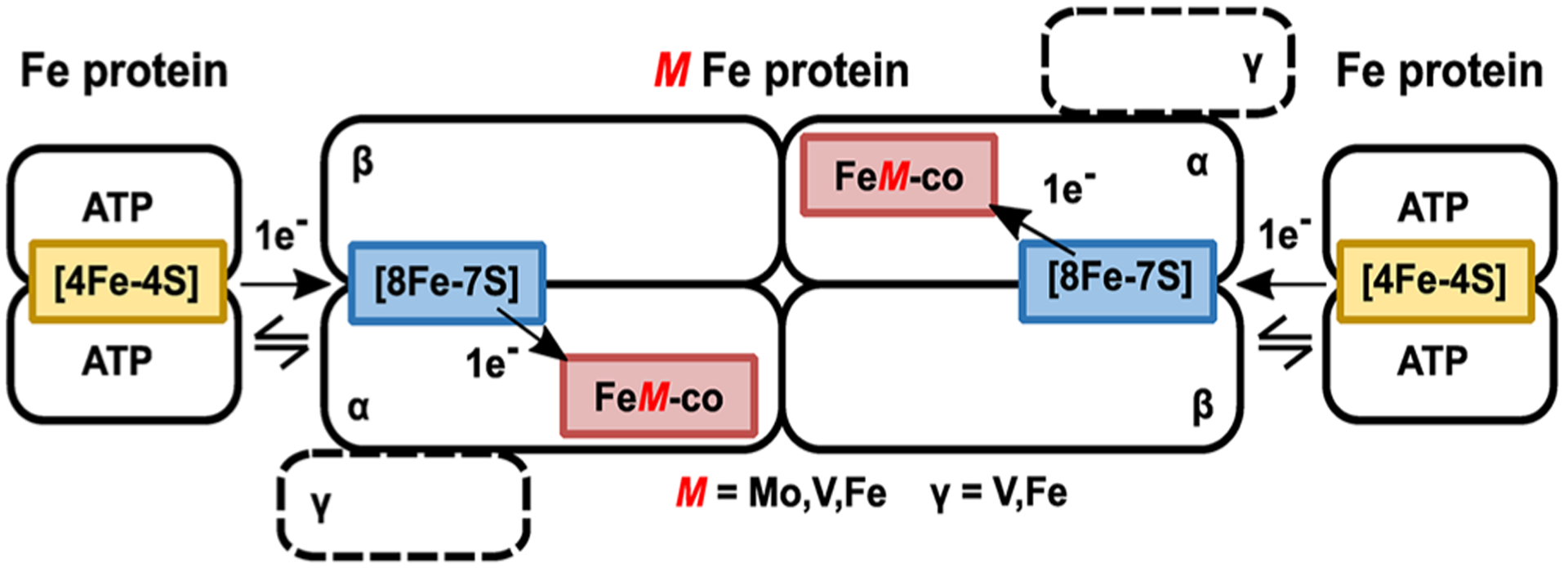
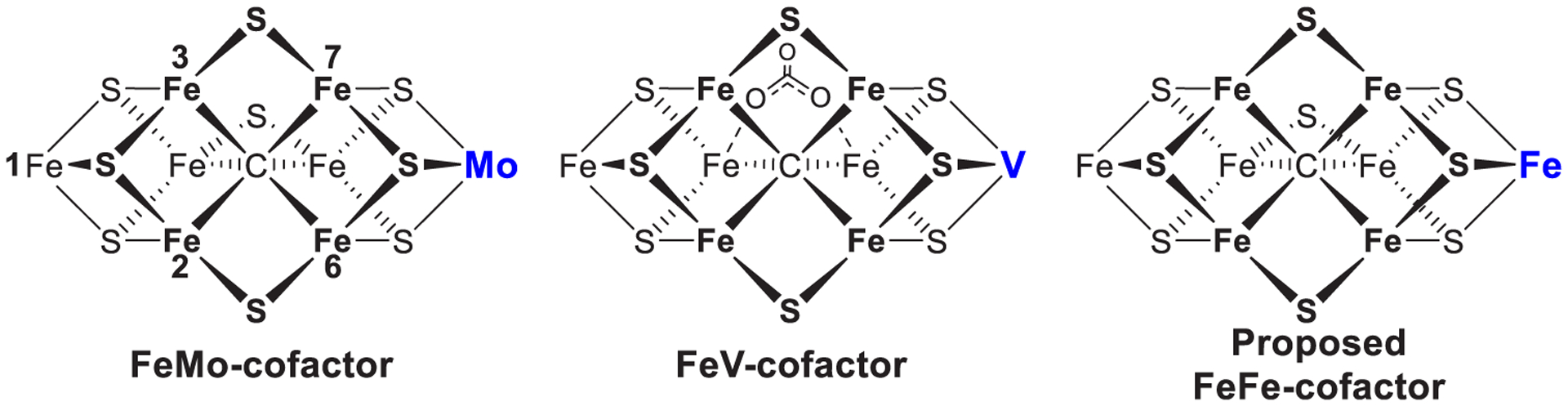
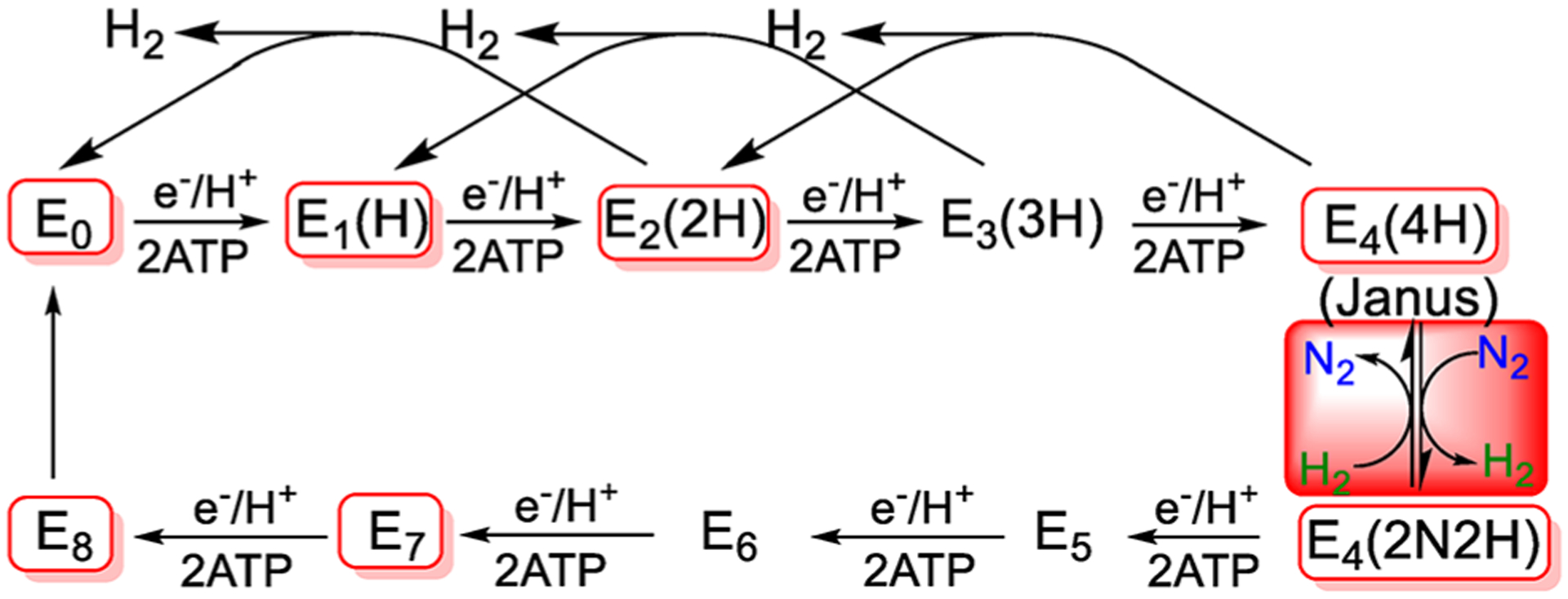
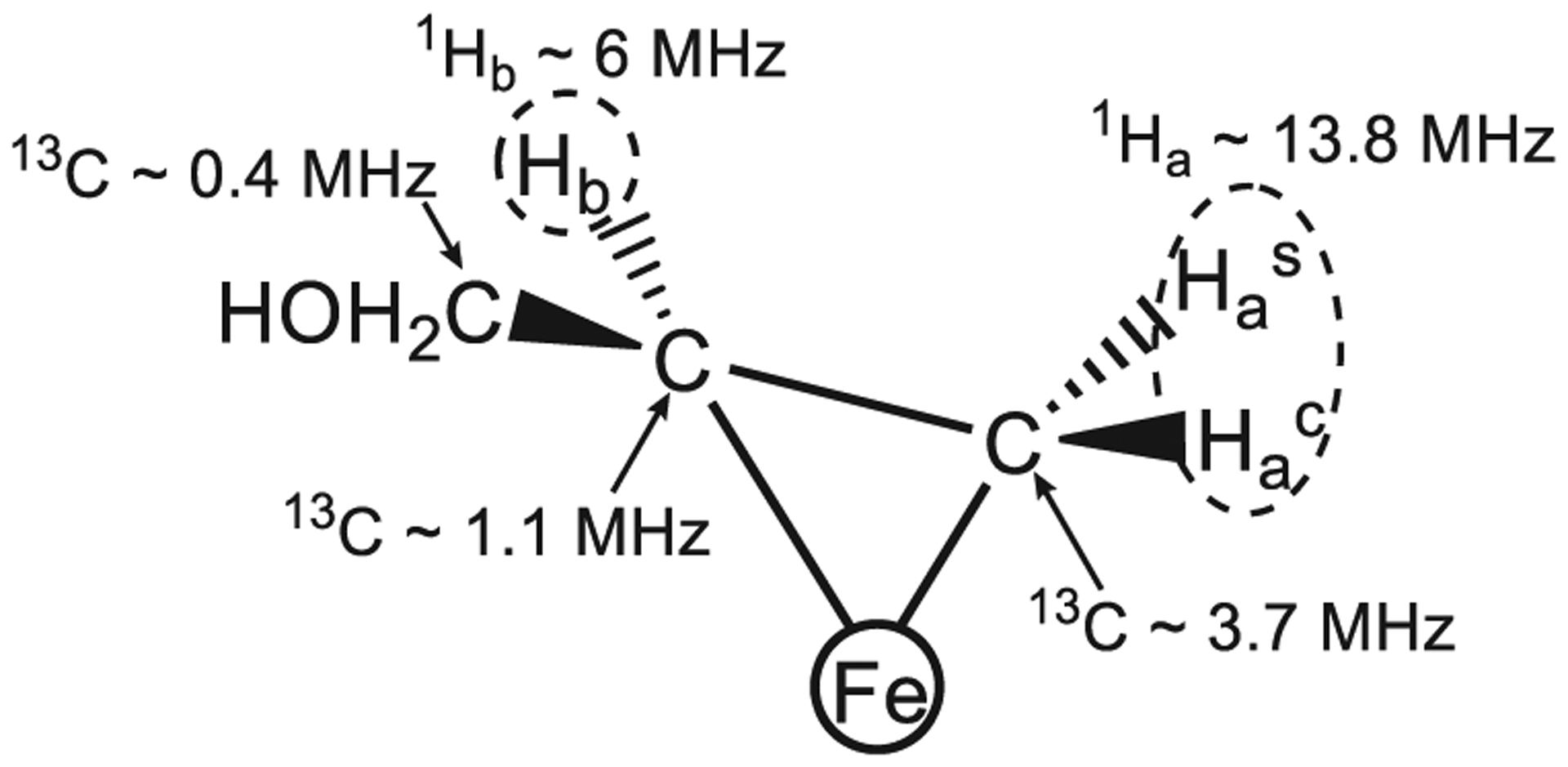
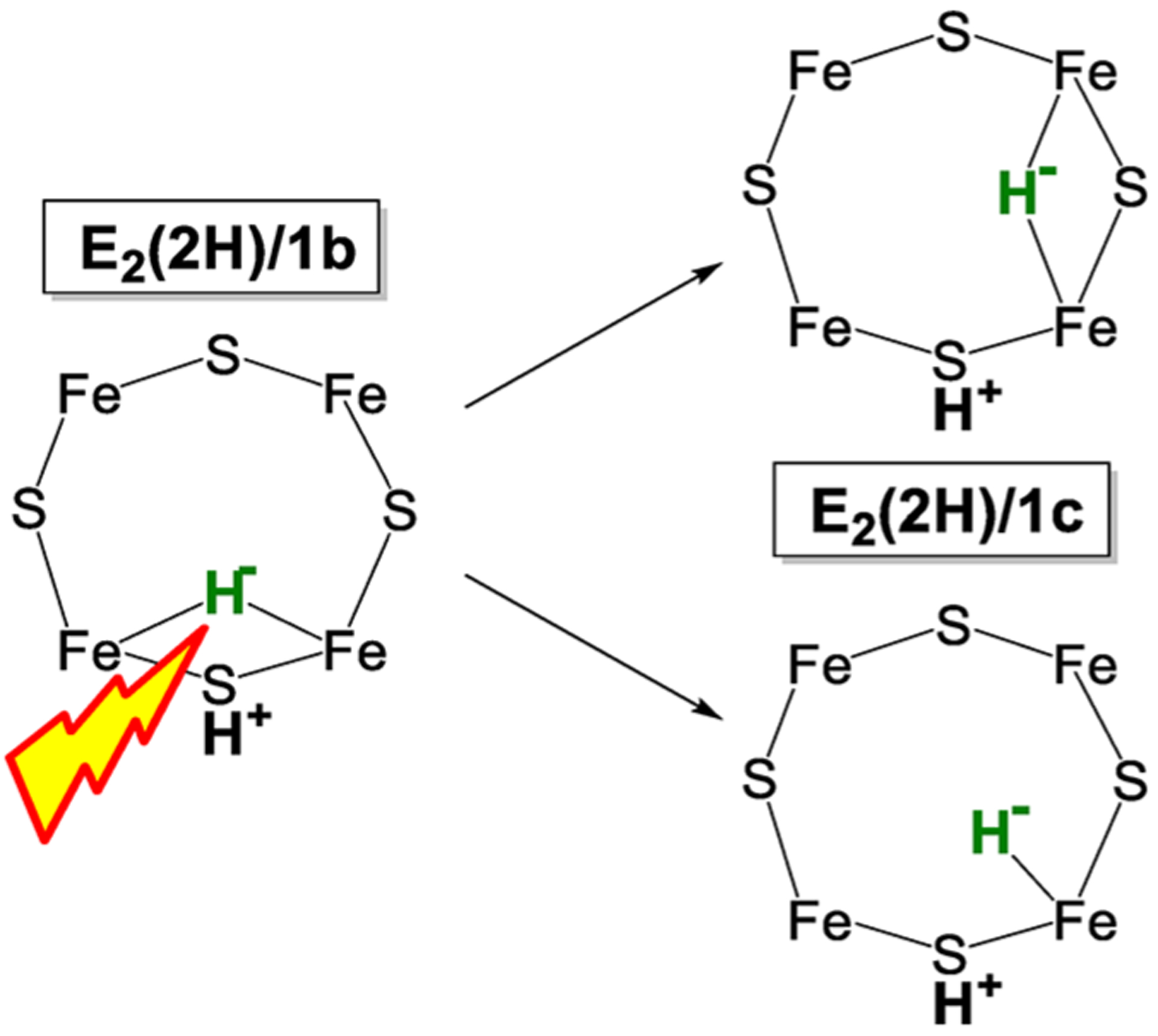
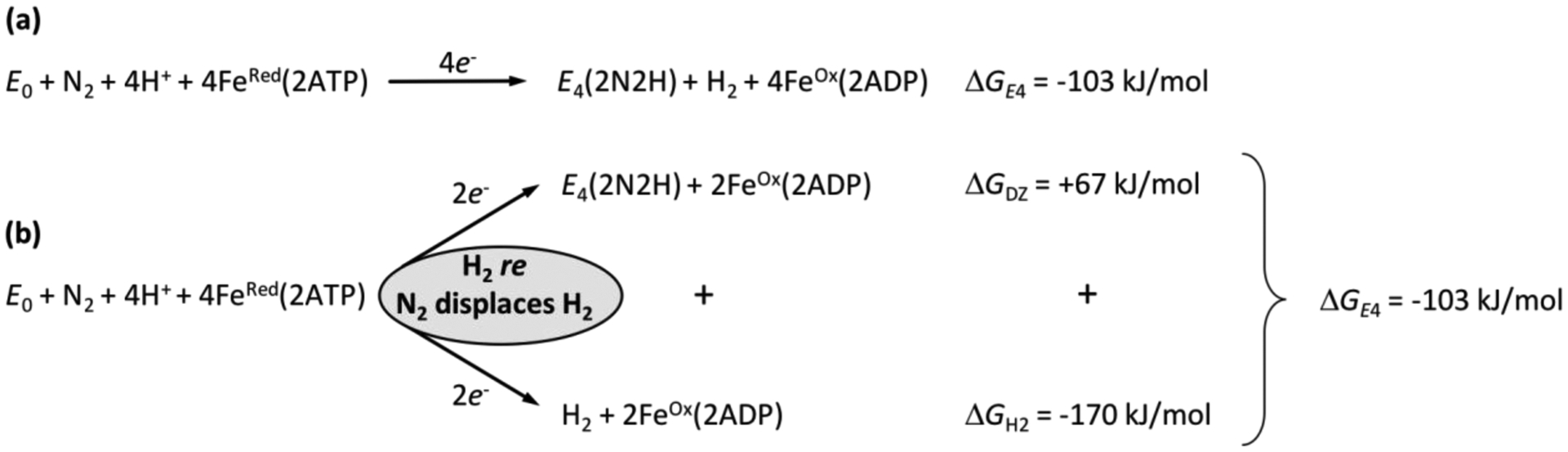
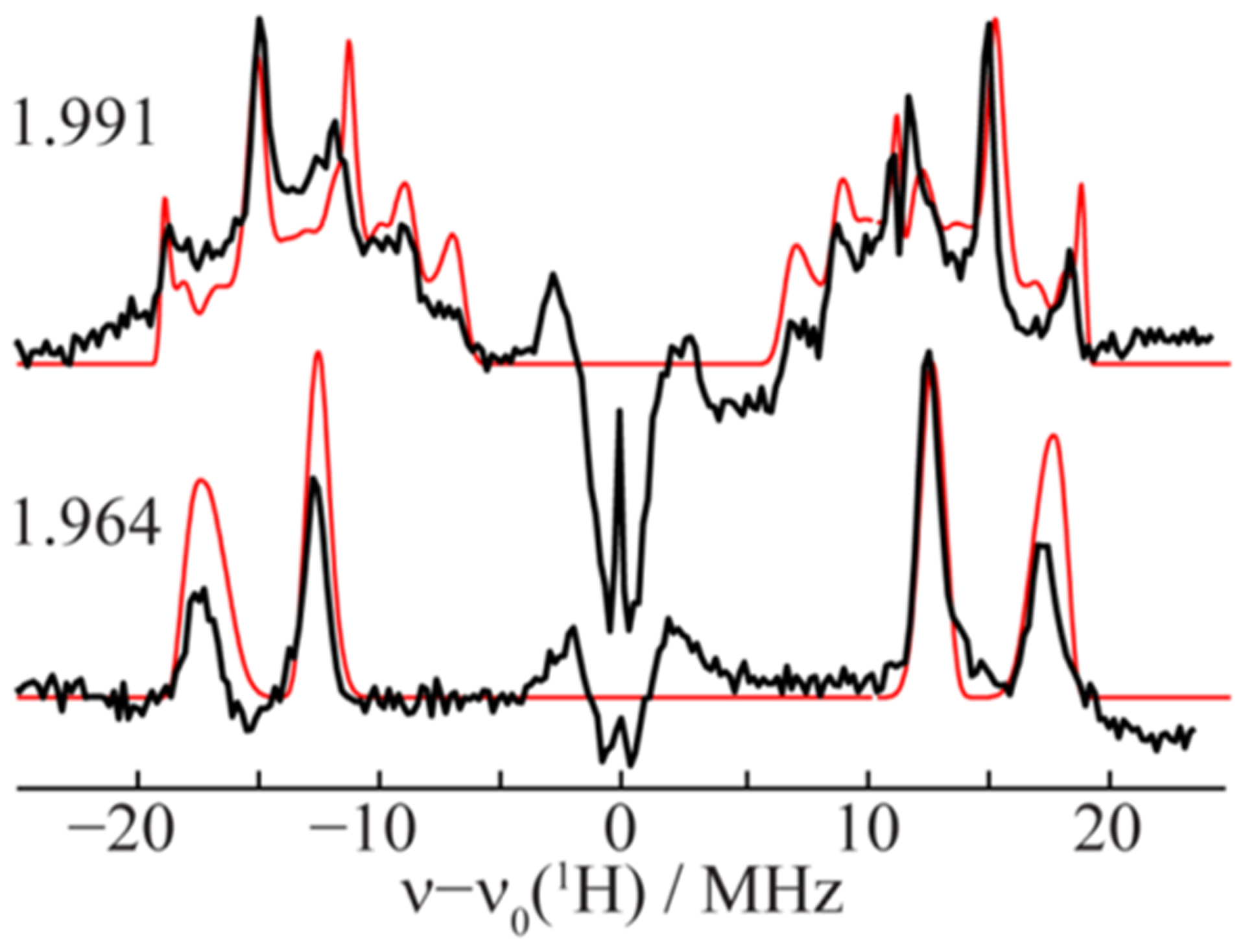
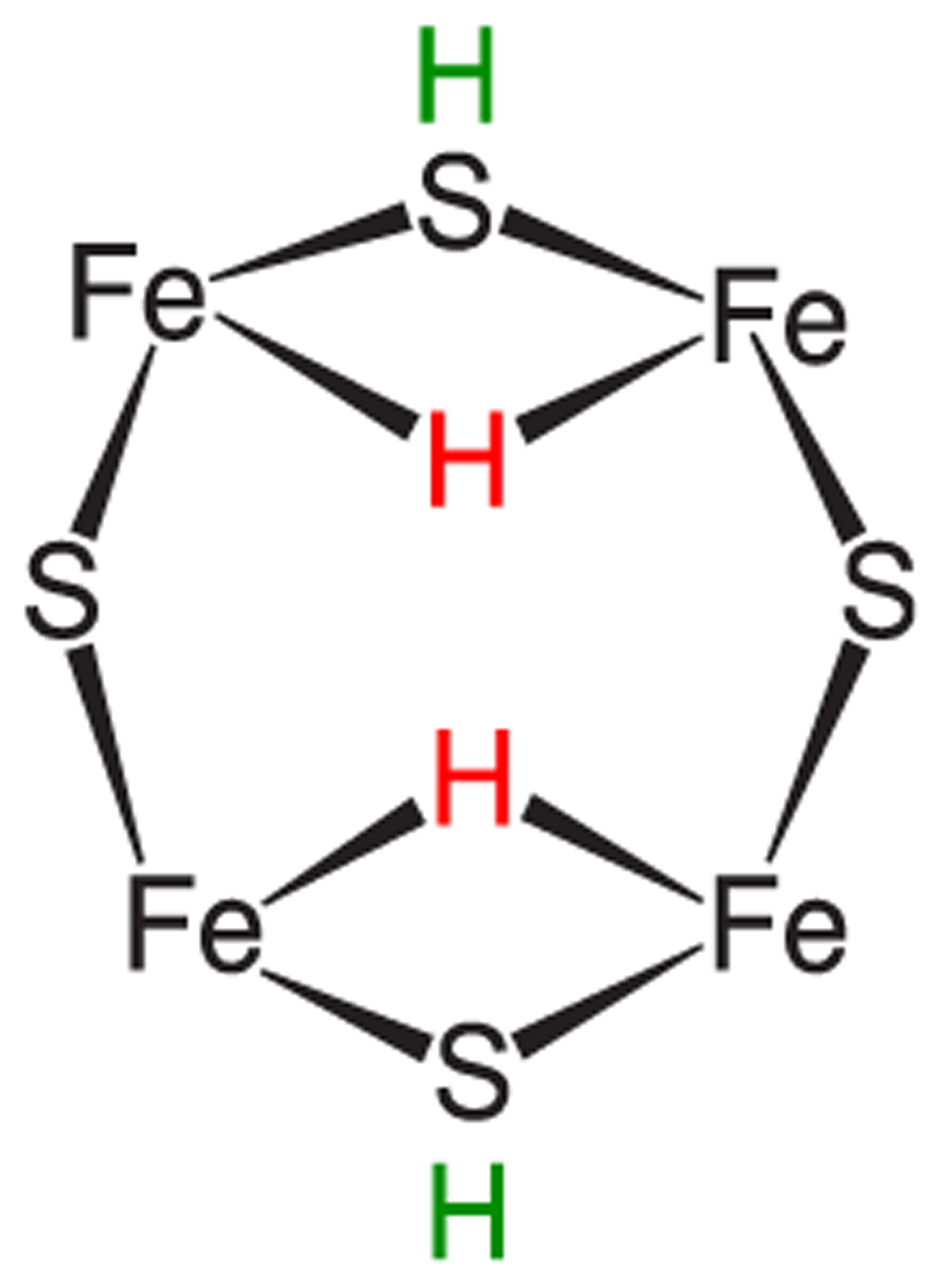
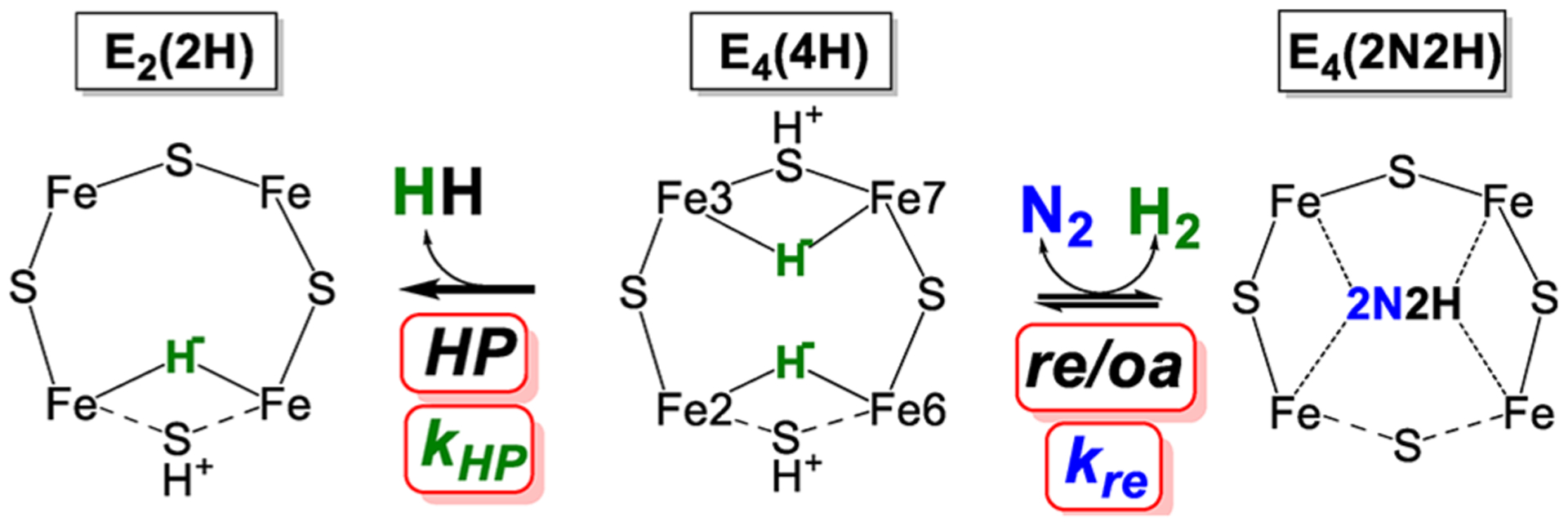
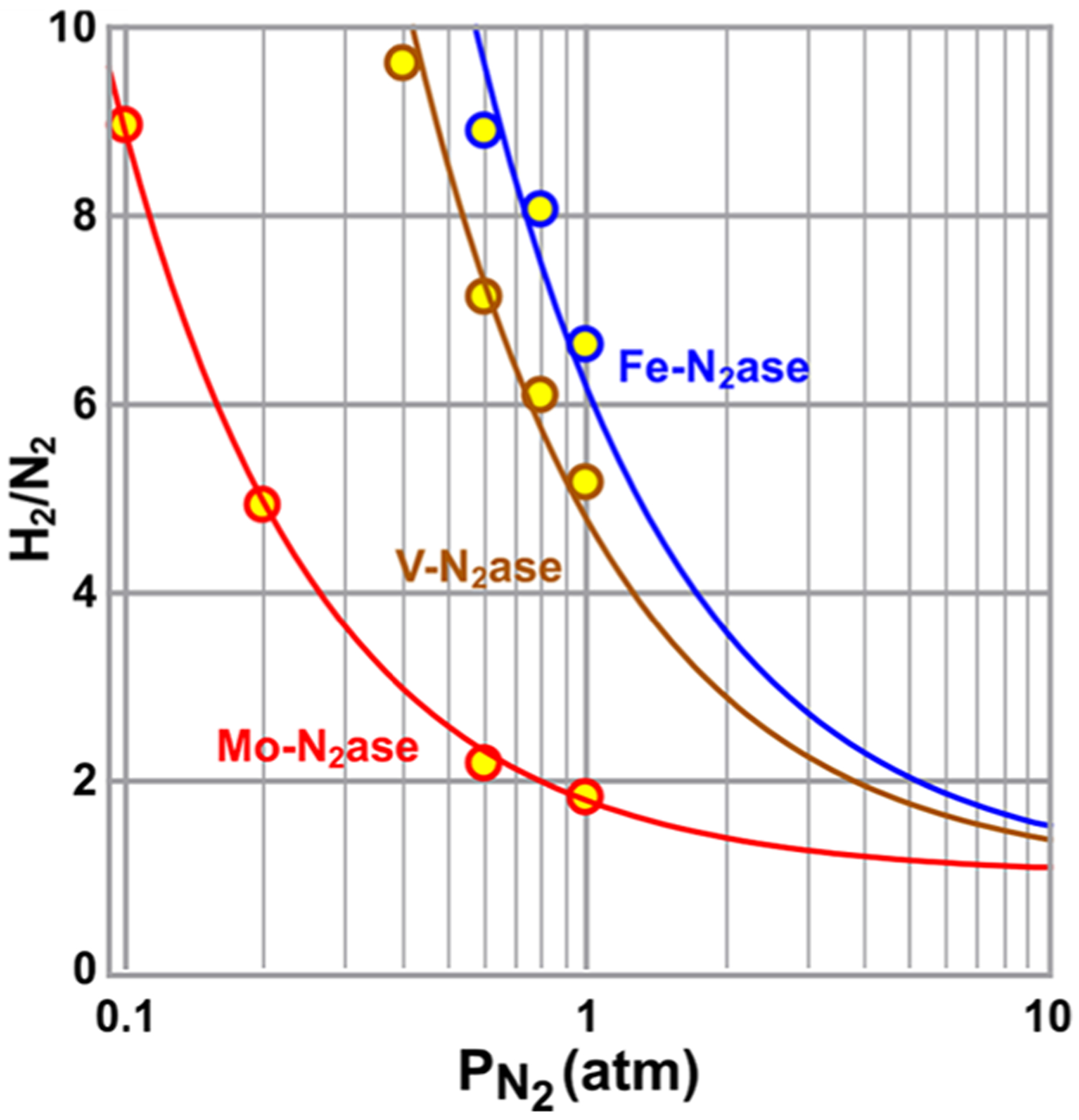
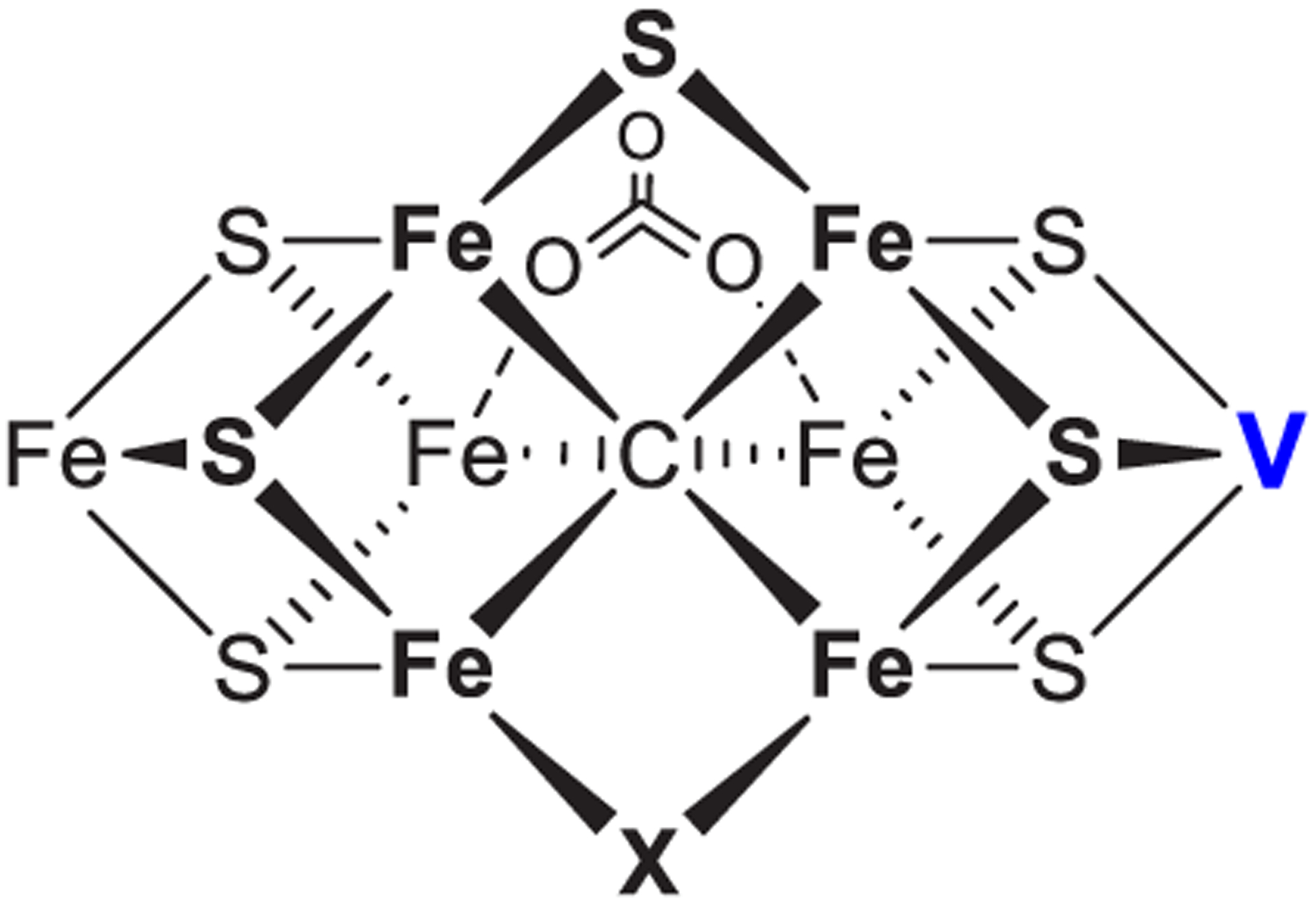
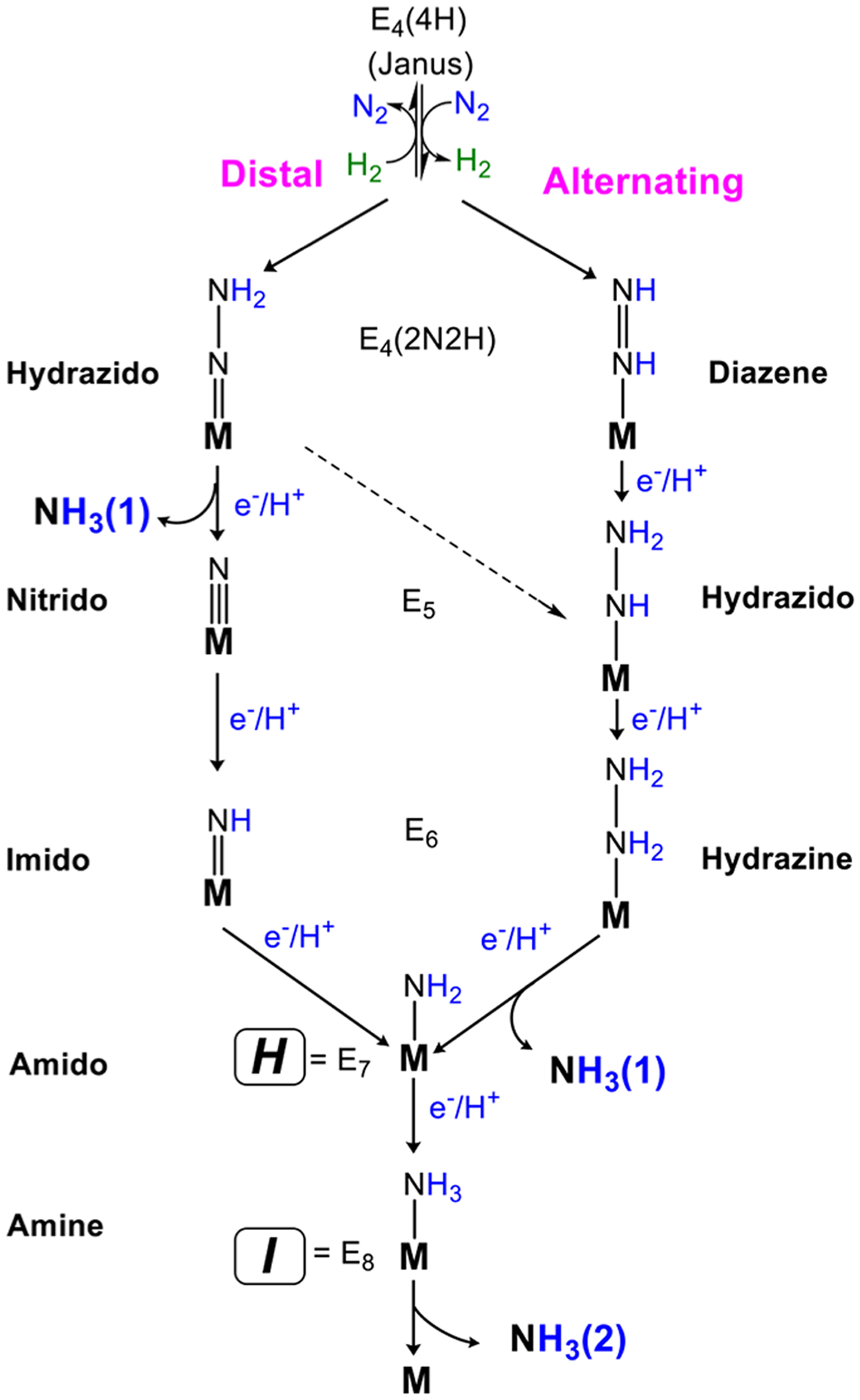
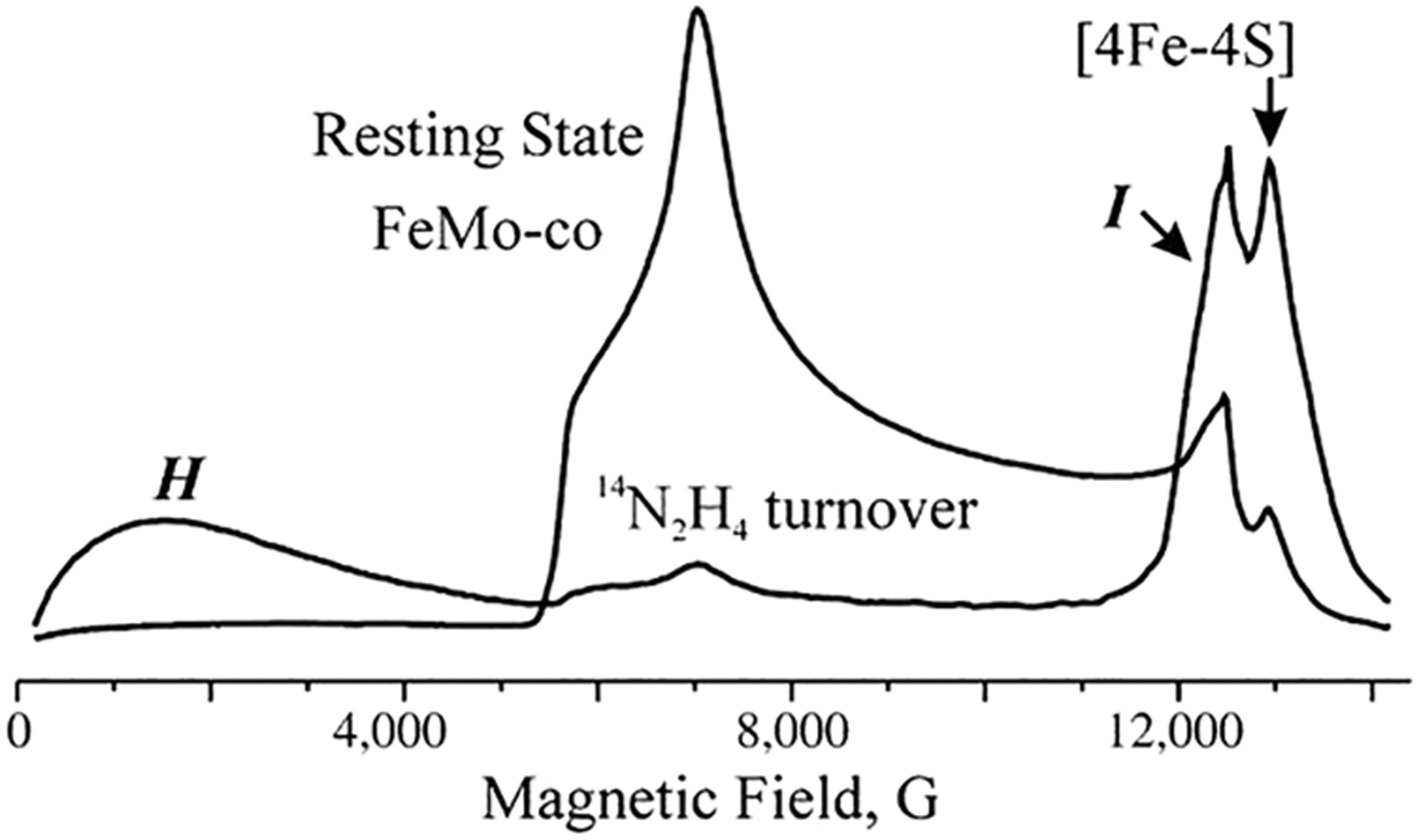
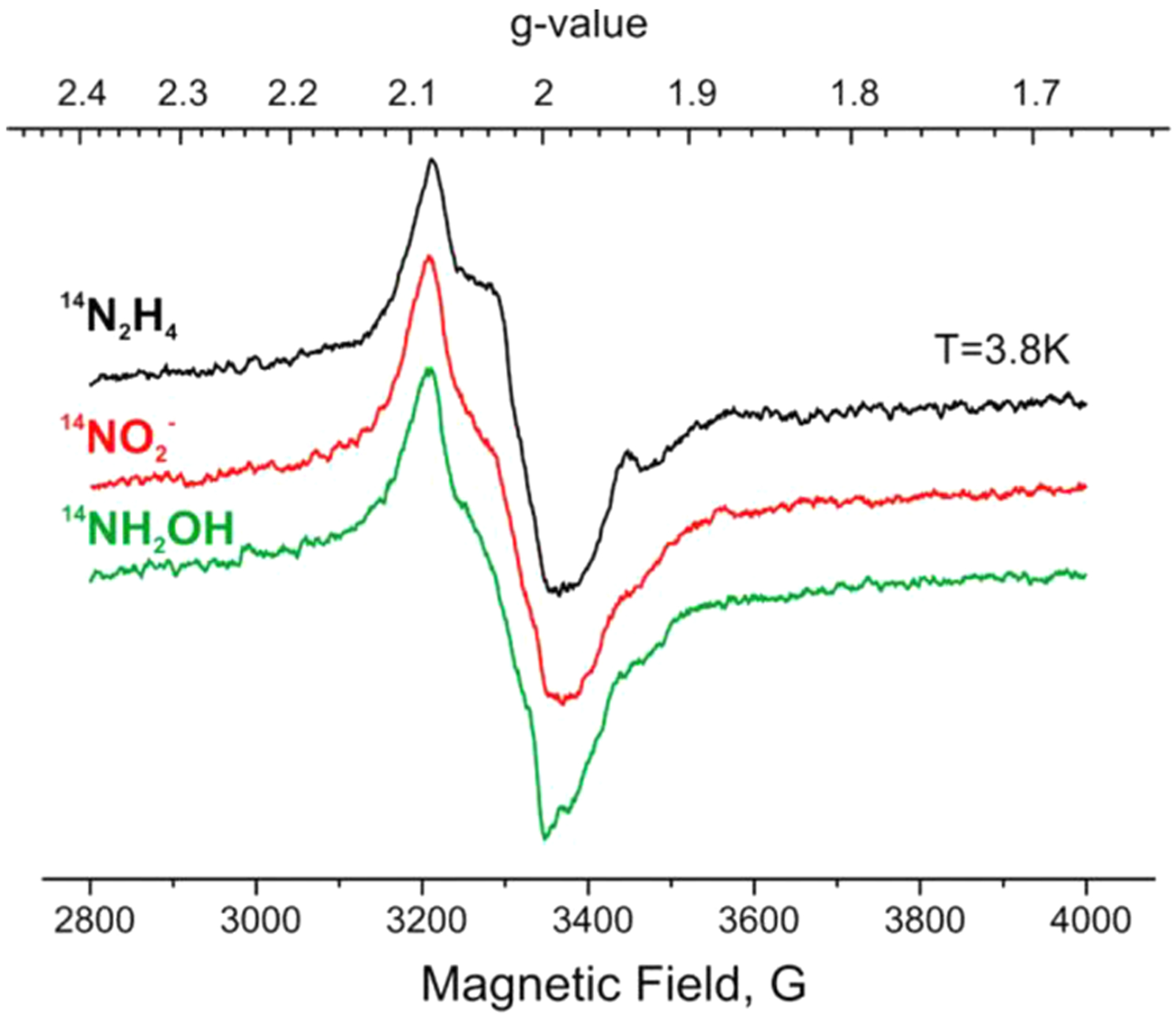
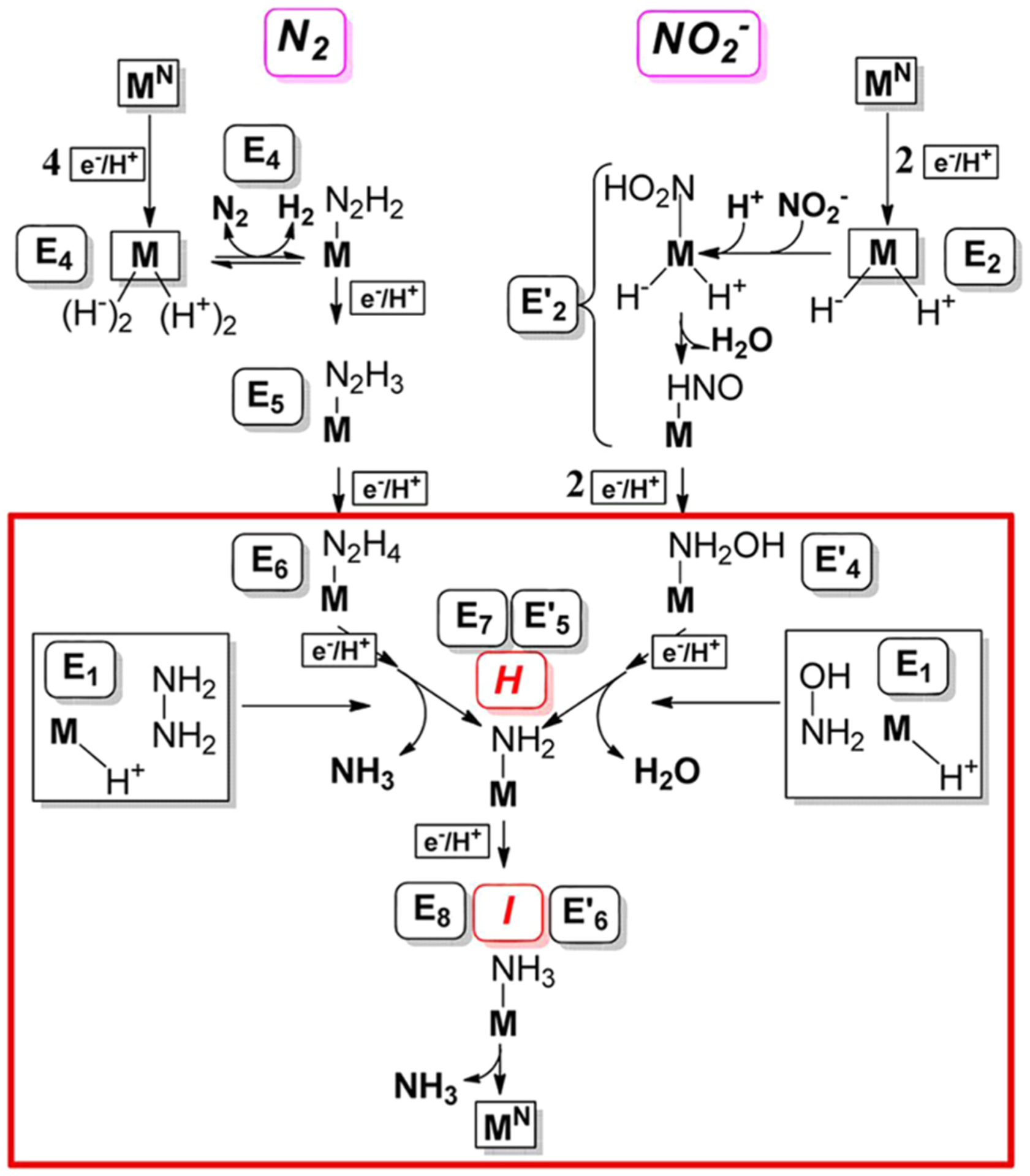
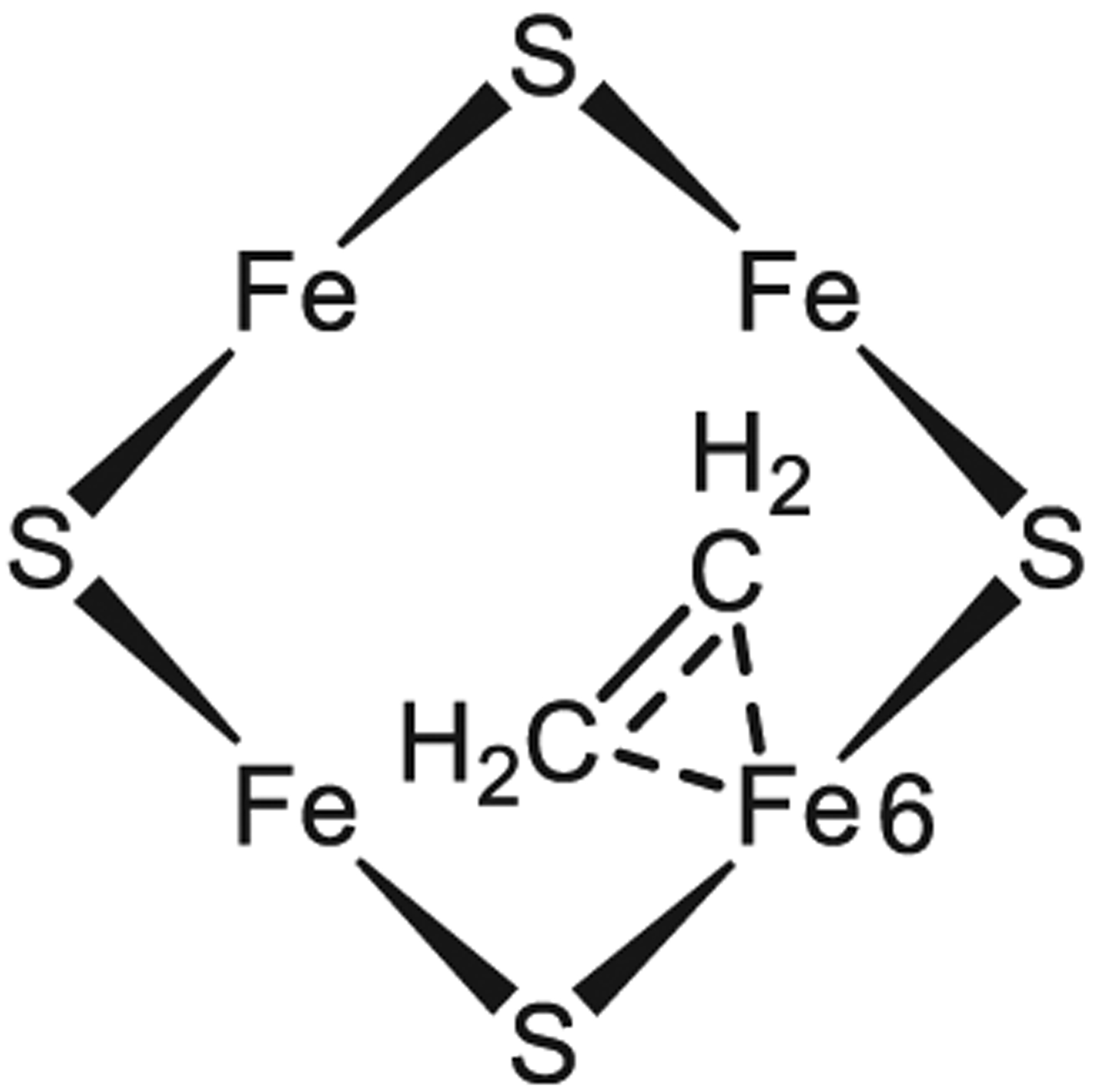
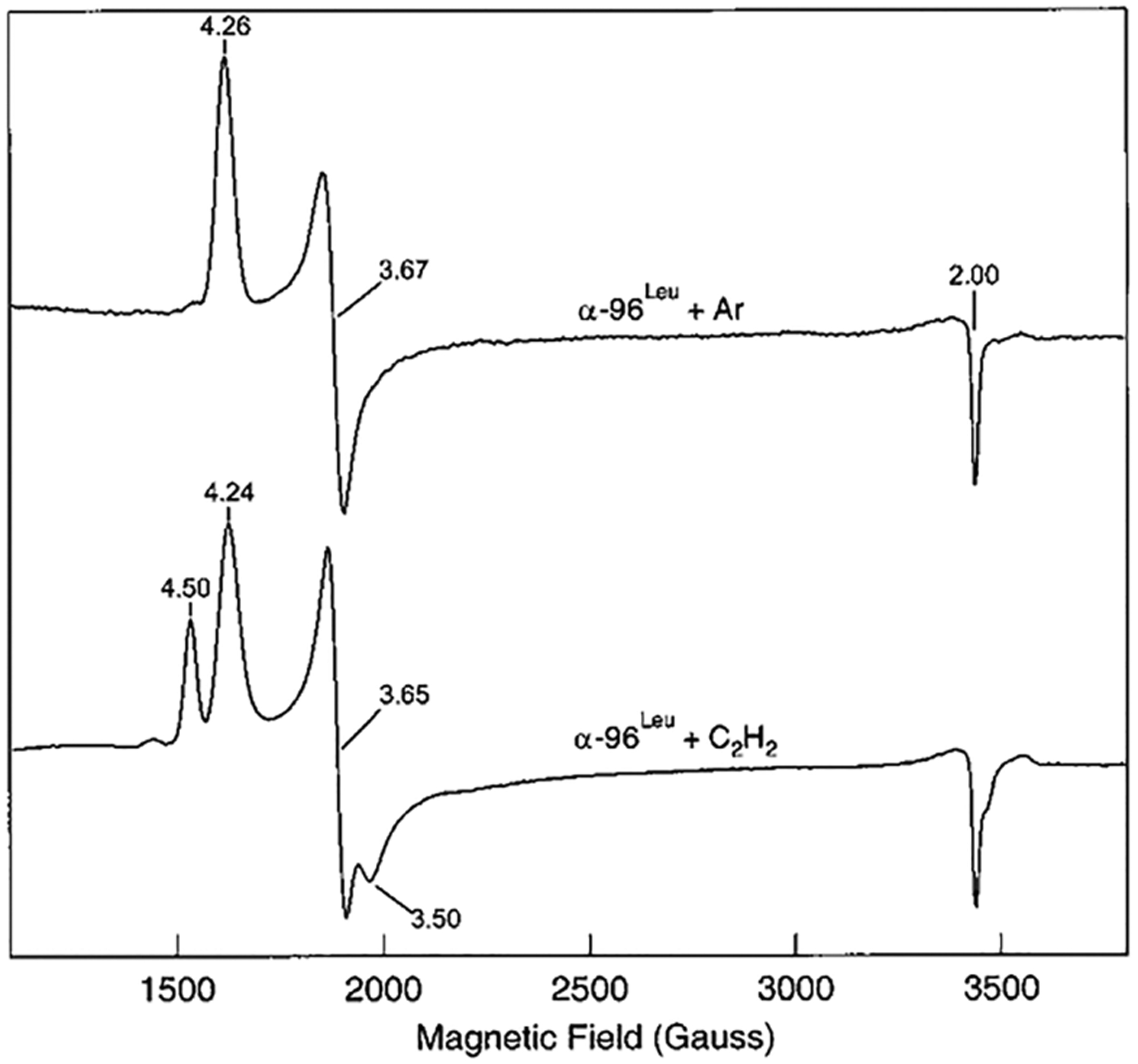
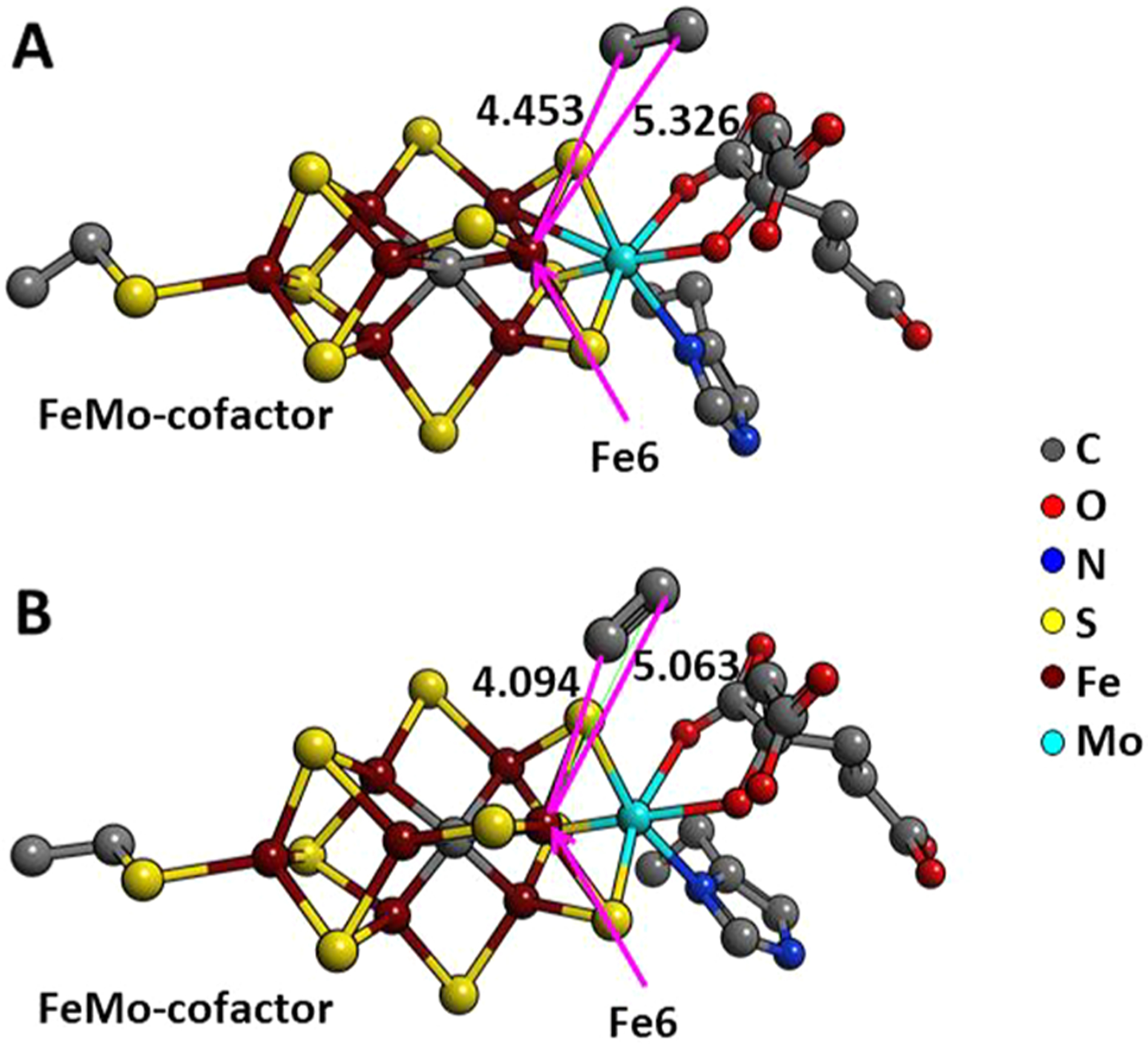
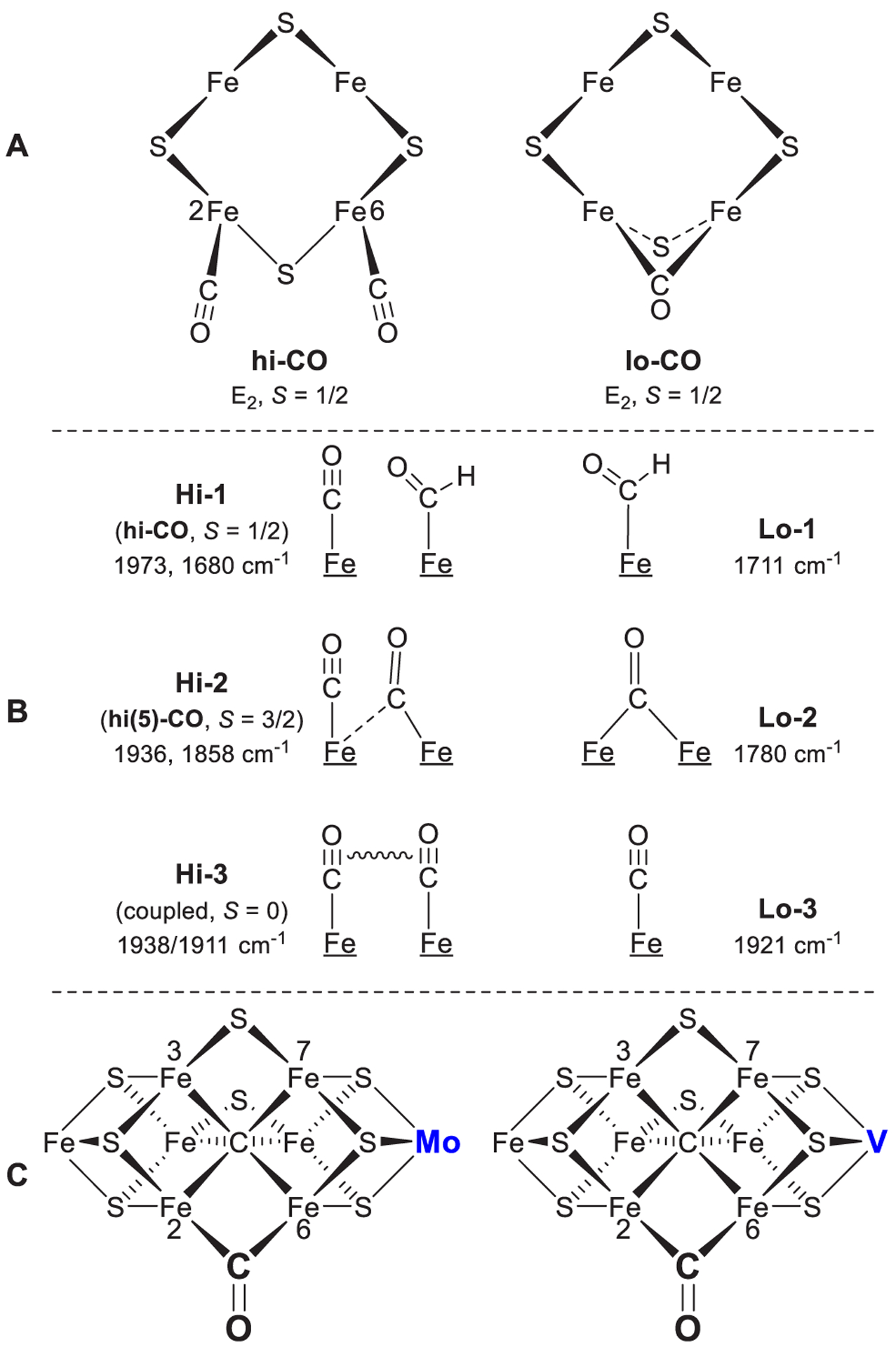
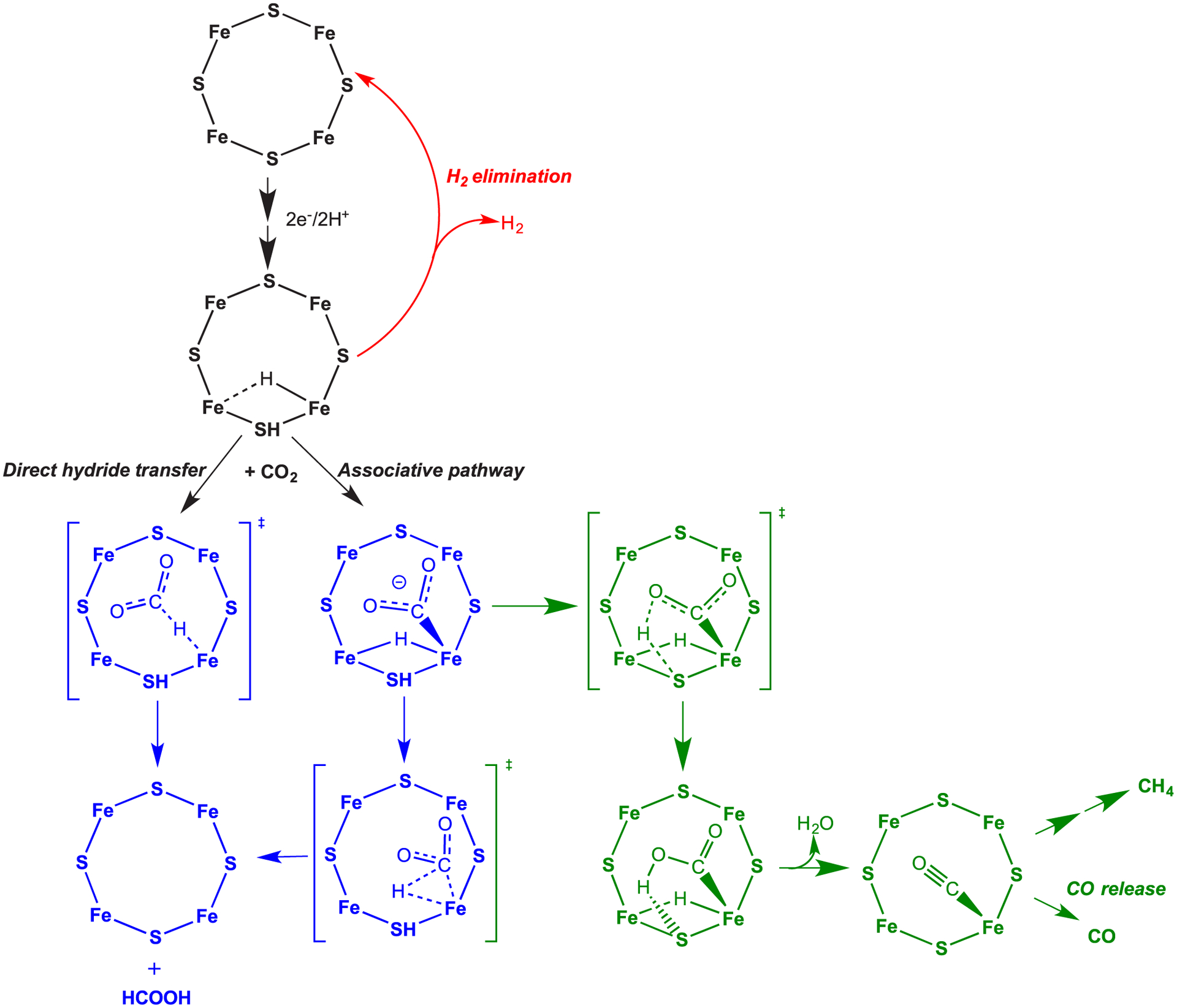
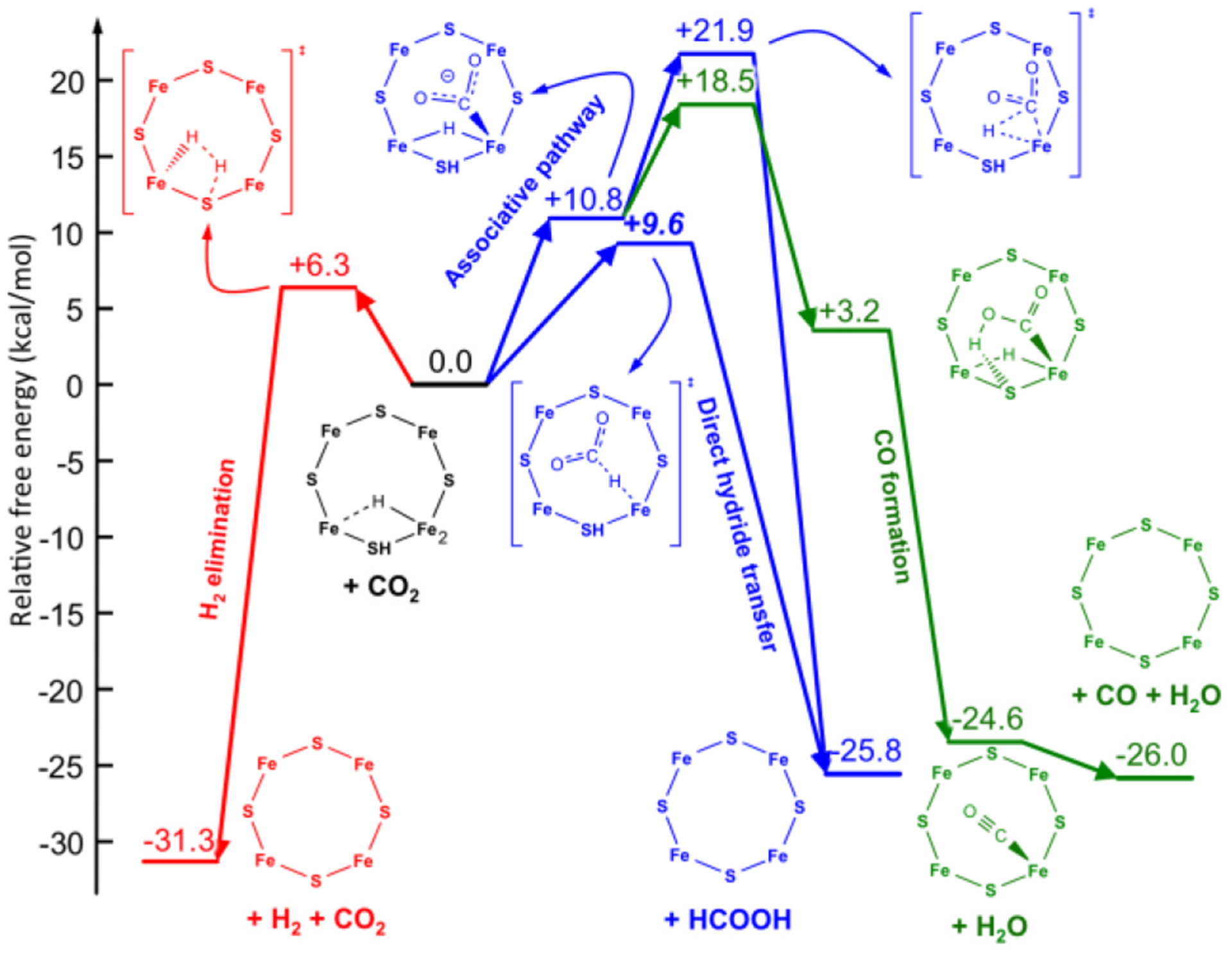
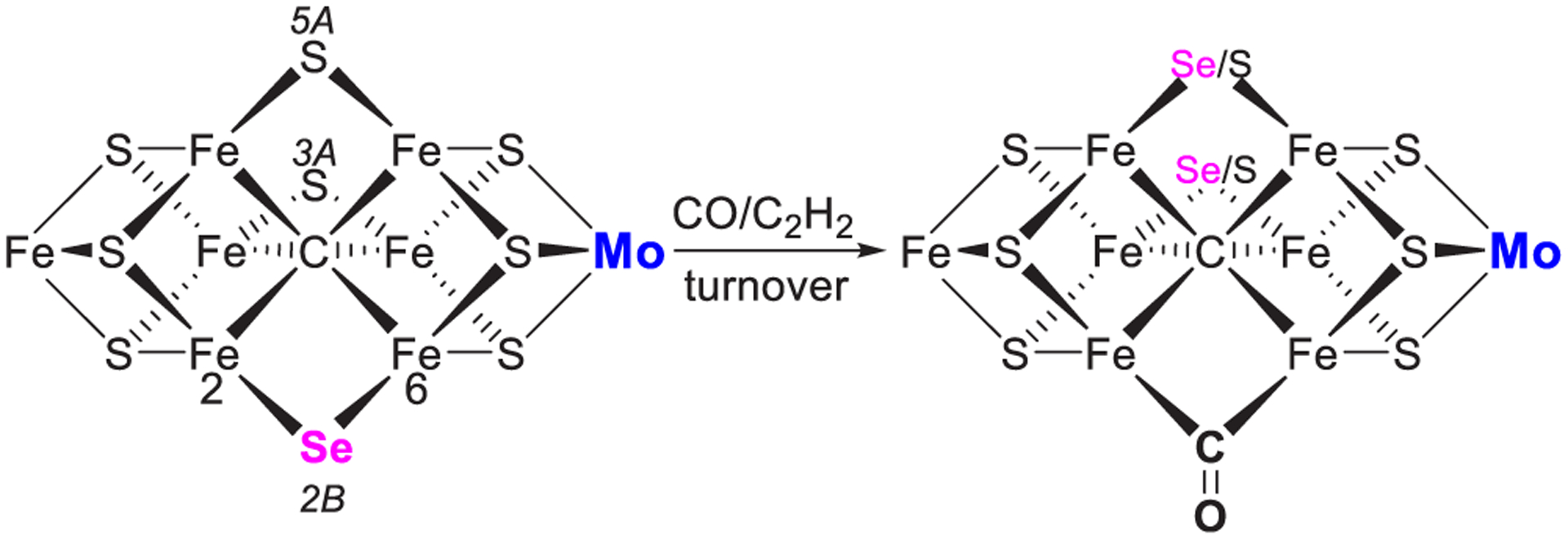

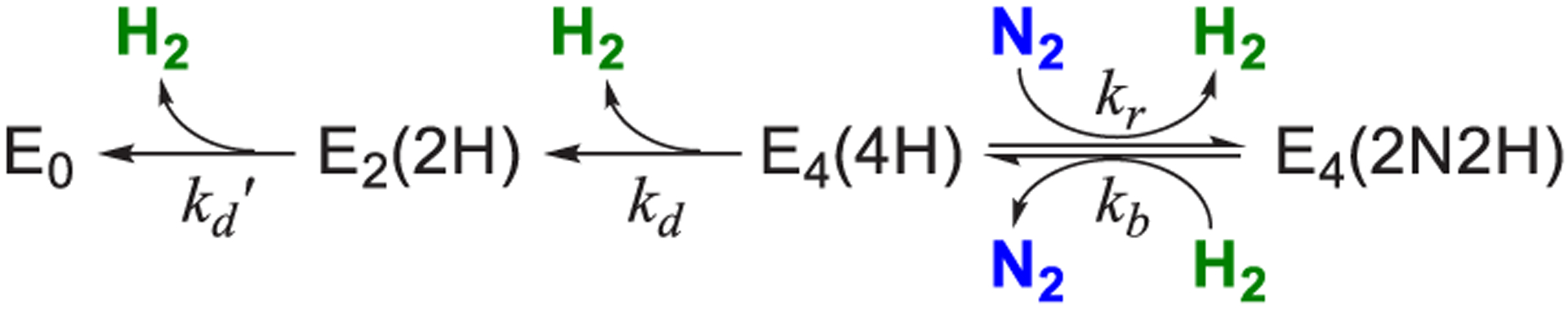
References
-
- Smil V Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production; MIT Press: Cambridge, MA, 2004.
-
- Erisman JW; Galloway JN; Dise NB; Sutton MA; Bleeker A; Grizzetti B; Leach AM; de Vries W Nitrogen: Too Much of a Vital Resource; Science Brief; WWF Nederland, 2015.
-
- Dawson CJ; Hilton J Fertiliser Availability in a Resource-Limited World: Production and Recycling of Nitrogen and Phosphorus. Food Policy 2011, 36, S14–S22.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
