

Review
doi: 10.1146/annurev-biochem-011520-104722.
Epub 2020 Mar 16.
Checkpoint Responses to DNA Double-Strand Breaks
Affiliations
- PMID: 32176524
- PMCID: PMC7311309
- DOI: 10.1146/annurev-biochem-011520-104722
Item in Clipboard
Review
Checkpoint Responses to DNA Double-Strand Breaks
Annu Rev Biochem.
.
Abstract
Cells confront DNA damage in every cell cycle. Among the most deleterious types of DNA damage are DNA double-strand breaks (DSBs), which can cause cell lethality if unrepaired or cancers if improperly repaired. In response to DNA DSBs, cells activate a complex DNA damage checkpoint (DDC) response that arrests the cell cycle, reprograms gene expression, and mobilizes DNA repair factors to prevent the inheritance of unrepaired and broken chromosomes. Here we examine the DDC, induced by DNA DSBs, in the budding yeast model system and in mammals.
Keywords: DNA double-strand break; DNA repair; cell cycle; checkpoint; kinases.
Figures
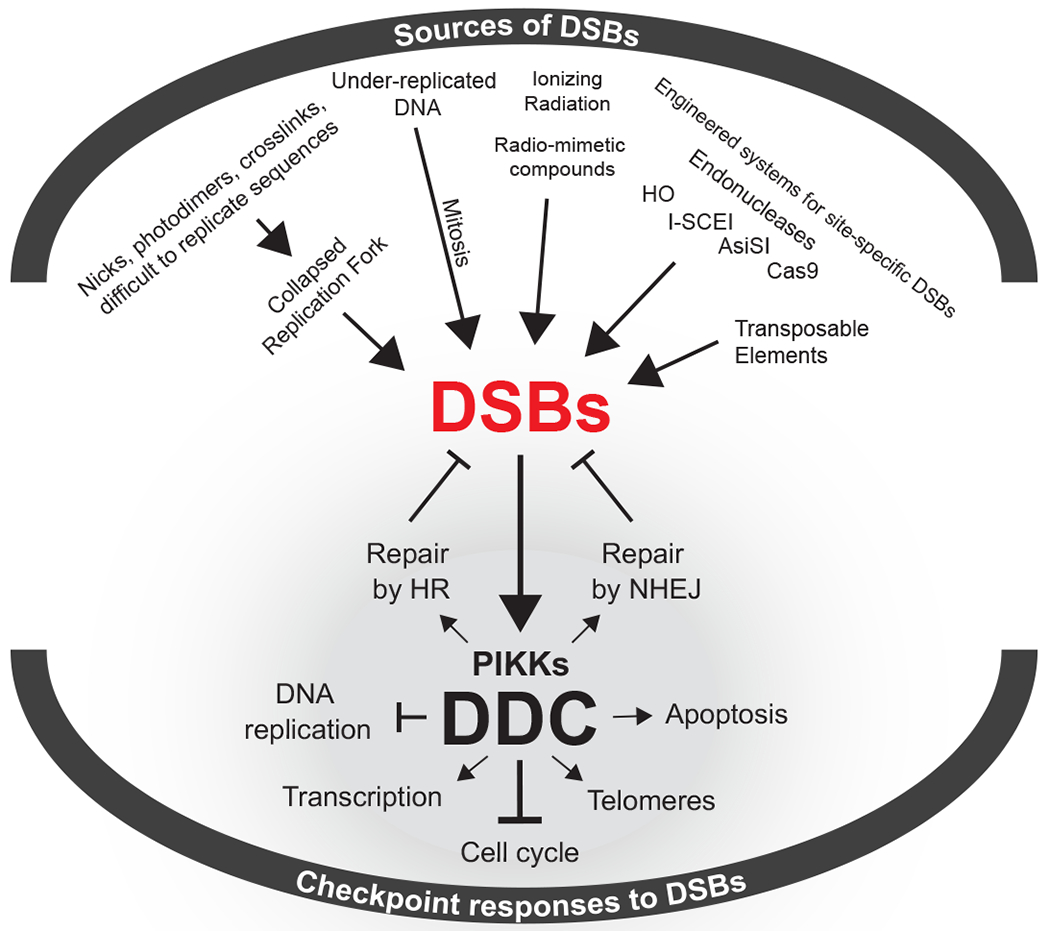
DSBs originating from endogenous or exogenous sources trigger the activation of DDC kinases that coordinate an intricate cellular response that includes cell cycle arrest and the mobilization of DSB-repair pathways.
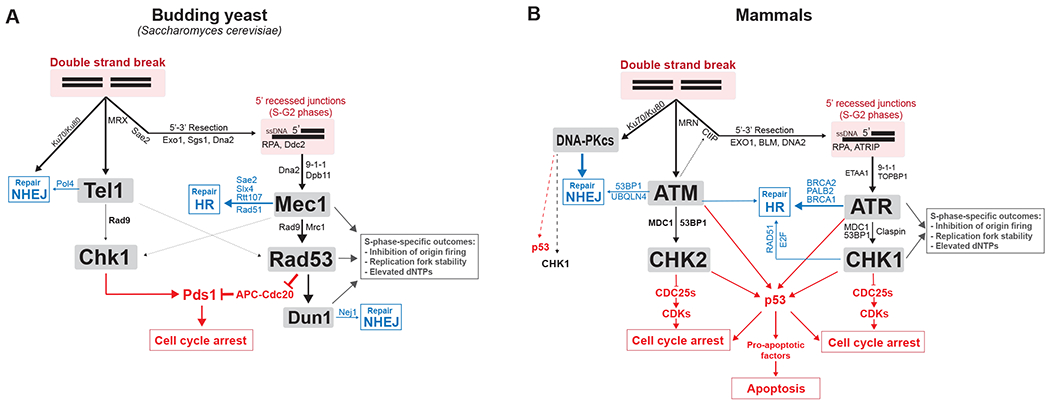
Simplified network view of the role of DDC kinases with their main co-factors, regulators, adaptors and substrates. Blue indicates key effectors through which DDC kinases regulate DNA repair of DSBs. Red indicates key effectors through which DDC kinases mediate cell cycle arrest and apoptosis. More information about substrates involved in DNA repair can be found here (197).
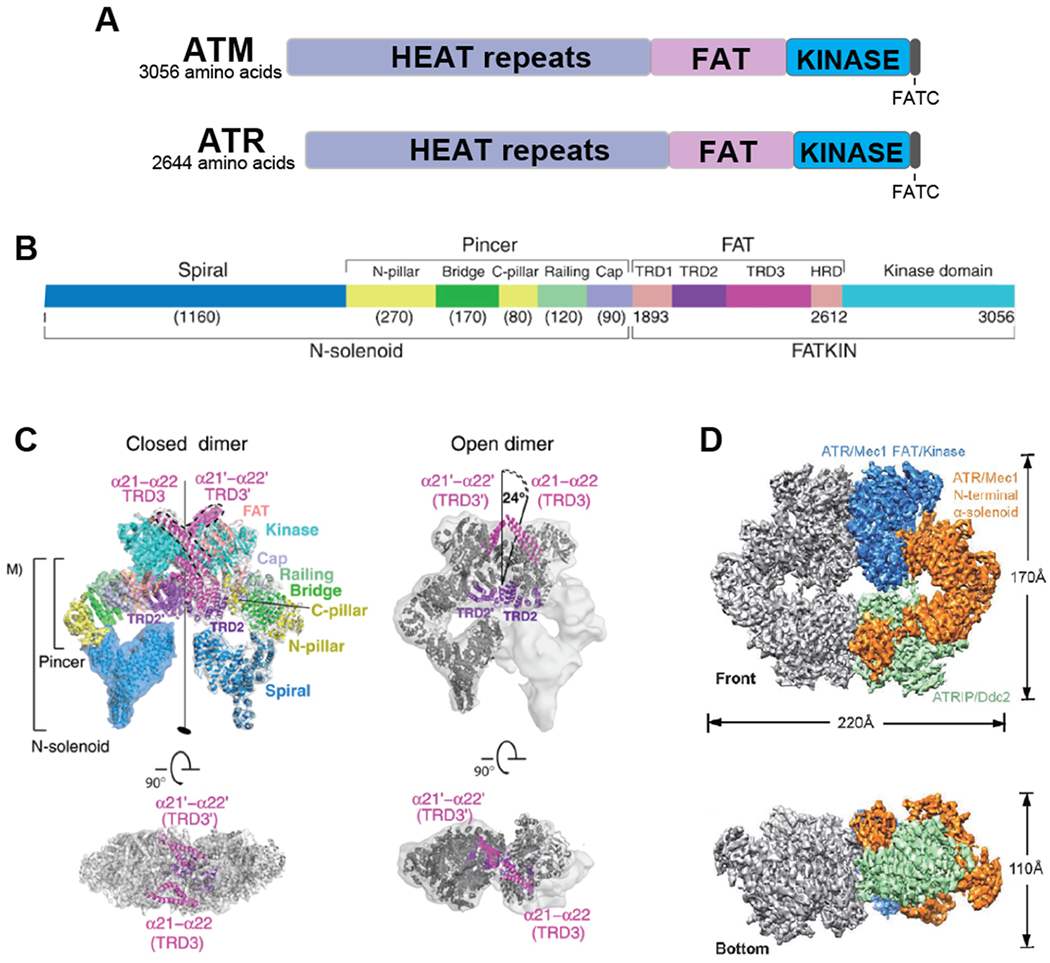
A. Overall domain structure of ATM and ATR. B. A more detailed structure of ATM including structural features identified by cryo-electron microscopy (45). C. Cryo-electron microscope-derived open (presumably active) and closed structures of ATM, as determined by Baretic et al. (45). The FAT and kinase region (FATKIN) has been solved at a higher resolution than the N-terminal solenoid domain. In the closed form, the conformation of the active site is maintained by interaction with a long helical hairpin in the TRD3 (tetratricopeptide repeats domain 3) domain. D. Electron microscope-derived structure of yeast Mec1-Ddc2 (ATR-ATRIP), determined by Wang et al. (47).
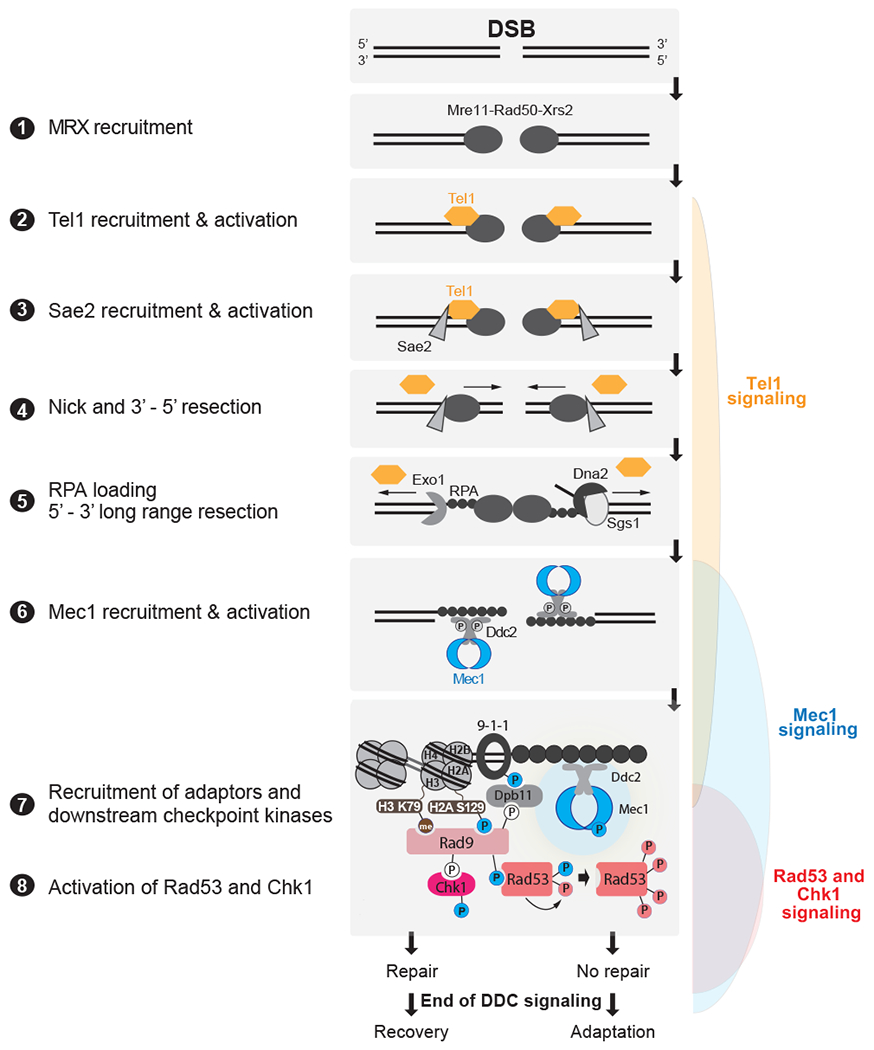
DSBs are first recognized by the MRX complex, which binds to the broken ends. MRX recruits the PIK kinase Tel1 and Sae2 to begin DSB end processing by Exo1 and Dna2. RPA coats ssDNA, leading to the recruitment of Mec1-Ddc2 dimers. The 9-1-1 clamp loader assembles the 9-1-1 clamp at the 5’ recessed end of the dsDNA/ssDNA junction. Dpb11 is recruited to 9-1-1 via a Mec1-dependent phosphorylation in the Ddc1 subunit. Mec1 and Tel1 phosphorylate histone H2A on S129 (Y-H2AX). Rad9 bound to Chkl is recruited to Y-H2AX through Rad9’s BRCT domain and histone H3K79me through Rad9’s TUDOR domain. Rad9 is shown here as monomeric for simplicity. Mec1 then phosphorylates and activates Chk1 and phosphorylates Rad9, priming Rad9 for Rad53 recruitment. Rad53 binds phosphorylated Rad9 allowing for Mec1-dependent phosphorylation and activation. Activated Rad53 phosphorylates and activates other Rad53 molecules leading to full checkpoint activation. Tel1 in yeast contributes a very modest role to activating Rad53 and Chk1. Bottom most panel shows key interactions and phosphorylation events involved in activation of the DDC. Color of phosphorylation sites (circles with “P”) refer to the kinase responsible: blue=Mec1, red=Rad53, white=CDK, gray=unknown. See text for additional details.
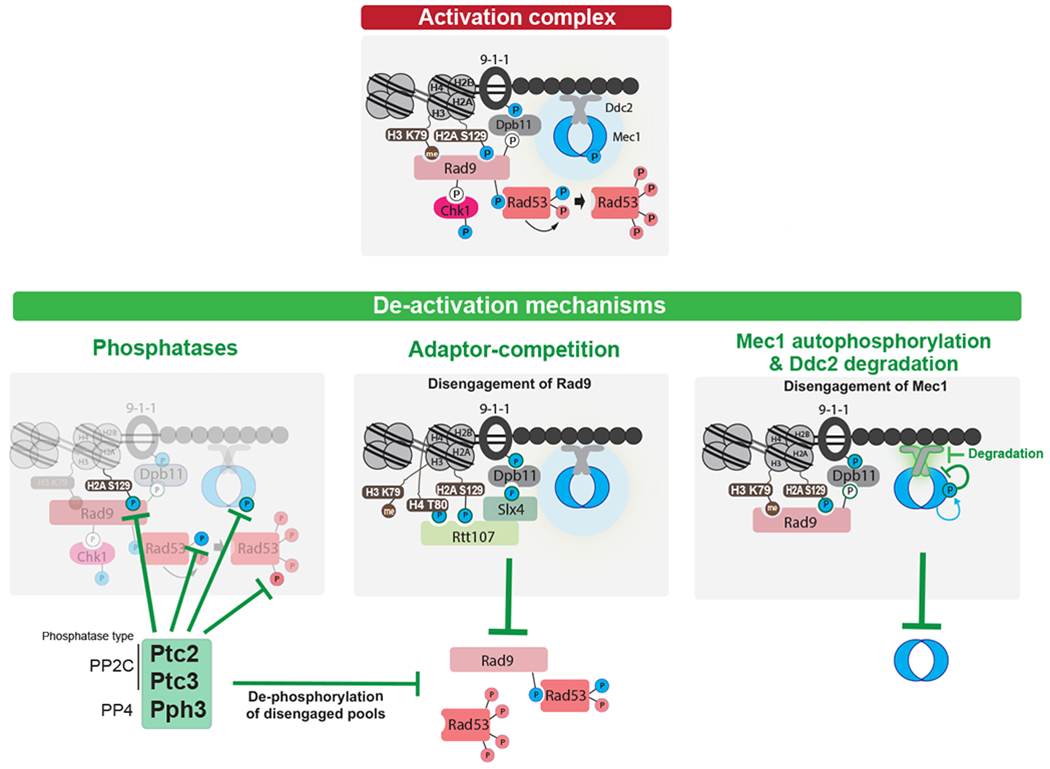
Top: Key interactions and phosphorylation events involved in activation of the DDC. Color of phosphorylation sites (circles with “P”) refer to the kinase responsible: blue=Mec1, red=Rad53, white=CDK. Bottom: three modes of down-regulating the DDC. Note that current evidence supports a model in which the PP4 phosphatase Pph3 removes Mec1 phosphorylation sites involved in Mec1 activation (237), while a later Mec1 autophosphorylation site triggers further Mec1 deactivation (210). For adaptor-competition, recent evidences suggest that Sae2 may also compete with Rad9 (238, 239), although the mechanism is not understood. See text for additional details.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
