

Inflammation in Traumatic Brain Injury
- PMID: 32176646
- PMCID: PMC8190673
- DOI: 10.3233/JAD-191150
Inflammation in Traumatic Brain Injury
Abstract
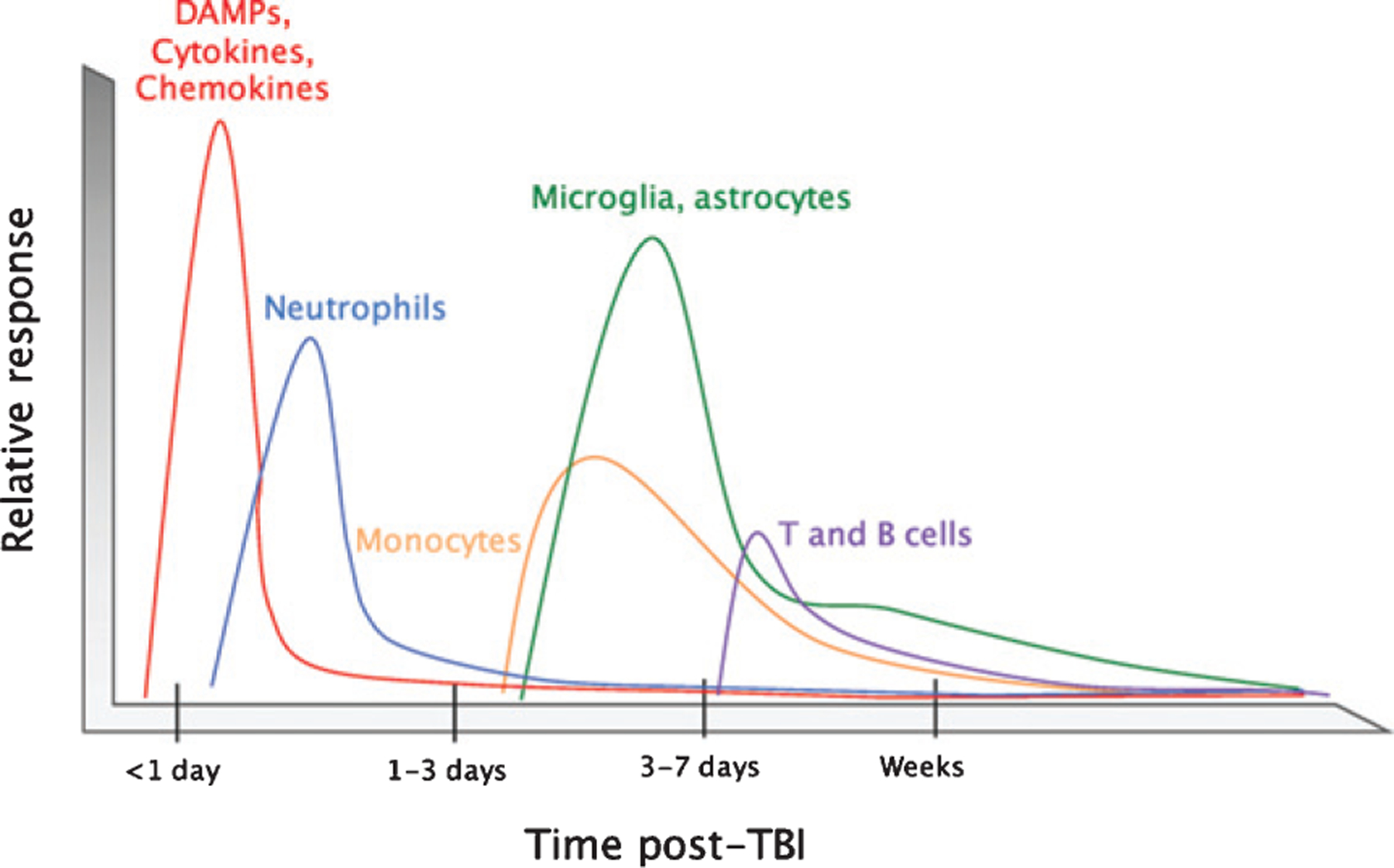
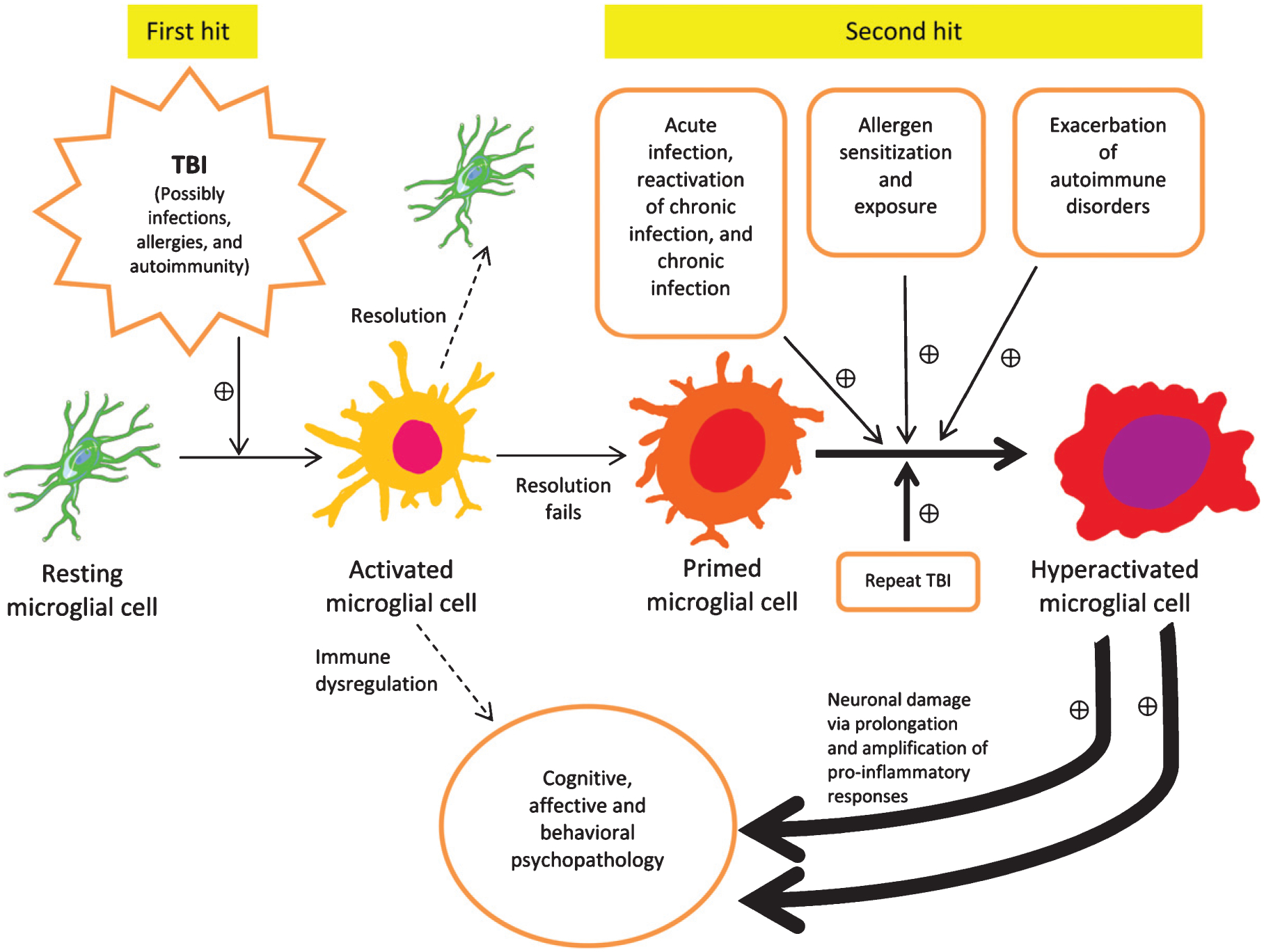
There is an increasing evidence that inflammation contributes to clinical and functional outcomes in traumatic brain injury (TBI). Many successful target-engaging, lesion-reducing, symptom-alleviating, and function-improving interventions in animal models of TBI have failed to show efficacy in clinical trials. Timing and immunological context are paramount for the direction, quality, and intensity of immune responses to TBI and the resulting neuroanatomical, clinical, and functional course. We present components of the immune system implicated in TBI, potential immune targets, and target-engaging interventions. The main objective of our article is to point toward modifiable molecular and cellular mechanisms that may modify the outcomes in TBI, and contribute to increasing the translational value of interventions that have been identified in animal models of TBI.
Keywords: Depression; glia; immune challenge; immunomodulation; inflammation; priming; probiotic; traumatic brain injury.
Figures

References
-
- Silver JM, McAllister TW, Arciniegas DB (2019) Textbook of traumatic brain injury. American Psychiatric Publishing.
-
- Moalem G, Leibowitz-Amit R, Yoles E, Mor F, Cohen IR, Schwartz M (1999) Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med 5, 49–55. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
