

Genetics of severe combined immunodeficiency
- PMID: 32181275
- PMCID: PMC7063414
- DOI: 10.1016/j.gendis.2019.07.004
Genetics of severe combined immunodeficiency
Abstract
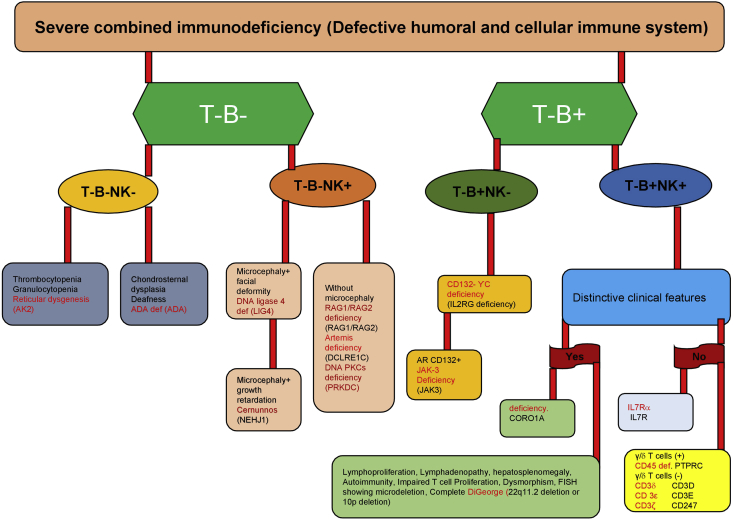
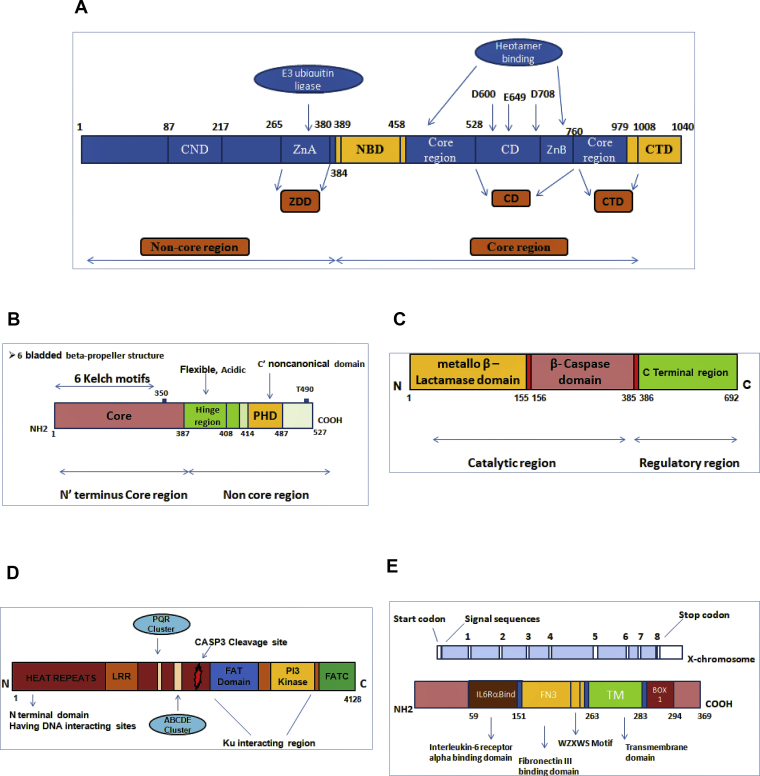
Severe Combined Immunodeficiency (SCID) is an inherited group of rare, life-threatening disorders due to the defect in T cell development and function. Clinical manifestations are characterised by recurrent and severe bacterial, viral, and fungal opportunistic infections that start from early infancy period. Haematopoietic stem cell transplantation (HSCT) is the treatment of choice. The pattern of inheritance of SCID may be X-linked or autosomal recessive. Though the diagnosis of SCID is usually established by flow cytometry-based tests, genetic diagnosis is often needed for genetic counselling, prognostication, and modification of pre-transplant chemotherapeutic agents. This review aims to highlight the genetic aspects of SCID.
Keywords: Adenosine deaminase; Flow cytometry; Genetics; Newborn screening; Severe combined immunodeficiency.
© 2019 Chongqing Medical University. Production and hosting by Elsevier B.V.
Figures
References
-
- Zago C.A., Jacob C.M., de Albuquerque Diniz E.M. Autoimmune manifestations in SCID due to IL-7Rmutations: Omenn syndrome and cytopenias. Hum Immunol. 2014;75(7):662–666. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
