

Facile Synthesis of a Stable Side-on Phosphinyne Complex by Redox Driven Intramolecular Cyclisation
- PMID: 32181544
- PMCID: PMC7540294
- DOI: 10.1002/chem.201905750
Facile Synthesis of a Stable Side-on Phosphinyne Complex by Redox Driven Intramolecular Cyclisation
Abstract
Alkyne complexes with vicinal substitution by a Lewis acid and a Lewis base at the coordinated alkyne are prospective frustrated Lewis pairs exhibiting a particular mutual distance and, hence, a specific activation potential. In this contribution, investigations on the generation of a WII alkyne complex bearing a phosphine as Lewis base and a carbenium group as Lewis acid are presented. Independently on potential substrates added, an intramolecular cyclisation product was always isolated. A subsequent deprotonation step led to an unprecedented side-on λ5 -phosphinyne complex, which is interpreted as highly zwitterionic according to visible absorption spectroscopy supported by TD-DFT. Low-temperature 31 P NMR and EPR spectroscopic measurements combined with time-dependent IR-spectroscopic monitoring provided insights in the mechanism of the cyclisation reaction. Decomposition of the multicomponent IR spectra by multivariate curve resolution and a kinetic hard-modelling approach allowed the derivation of kinetic parameters. Assignment of the individual IR spectra to potential intermediates was provided by DFT calculations.
Keywords: alkyne complex; cyclisation mechanism; frustrated Lewis pair; non-innocent ligand; phosphinyne complex.
© 2020 The Authors. Published by Wiley-VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


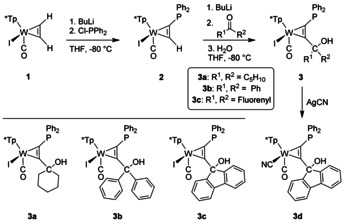


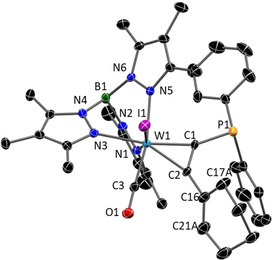
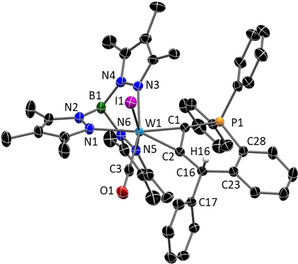










References
-
- None
-
- Yeoman J. T. S., Reisman S. E., Nature 2012, 490, 179–180; - PubMed
-
- Pozo I., Guitian E., Perez D., Pena D., Acc. Chem. Res. 2019, 52, 2472–2481; - PubMed
-
- Biehl E. R., Khanapure S. P., Acc. Chem. Res. 1989, 22, 275–281;
-
- Peña D., Pérez D., Guitián E., Angew. Chem. Int. Ed. 2006, 45, 3579–3581; - PubMed
- Angew. Chem. 2006, 118, 3659–3661;
Grants and funding
LinkOut - more resources
Full Text Sources
