

Glucocerebrosidase: Functions in and Beyond the Lysosome
- PMID: 32182893
- PMCID: PMC7141376
- DOI: 10.3390/jcm9030736
Glucocerebrosidase: Functions in and Beyond the Lysosome
Abstract
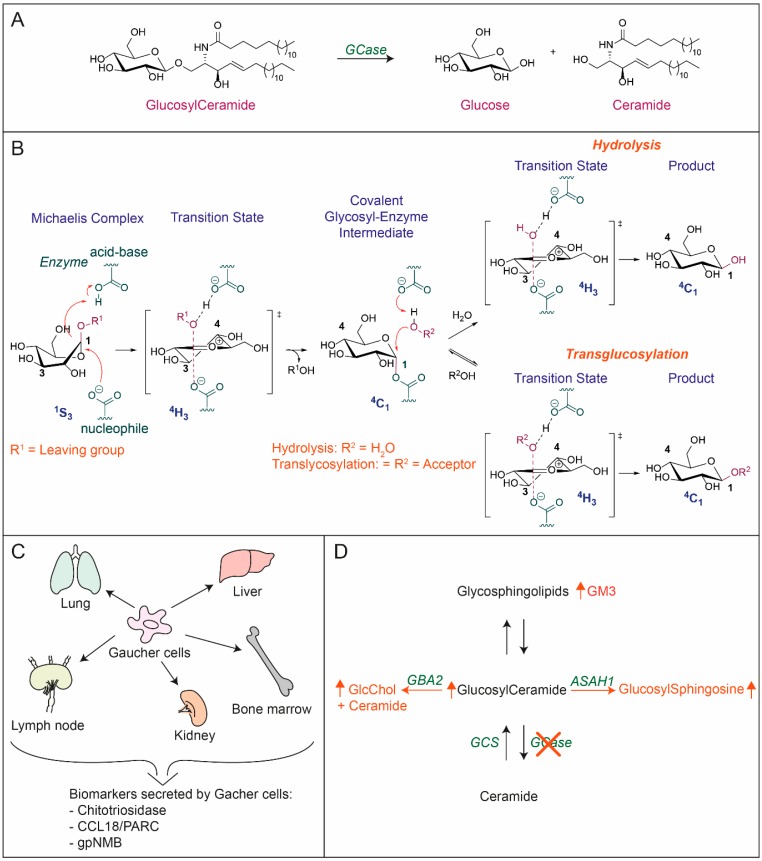
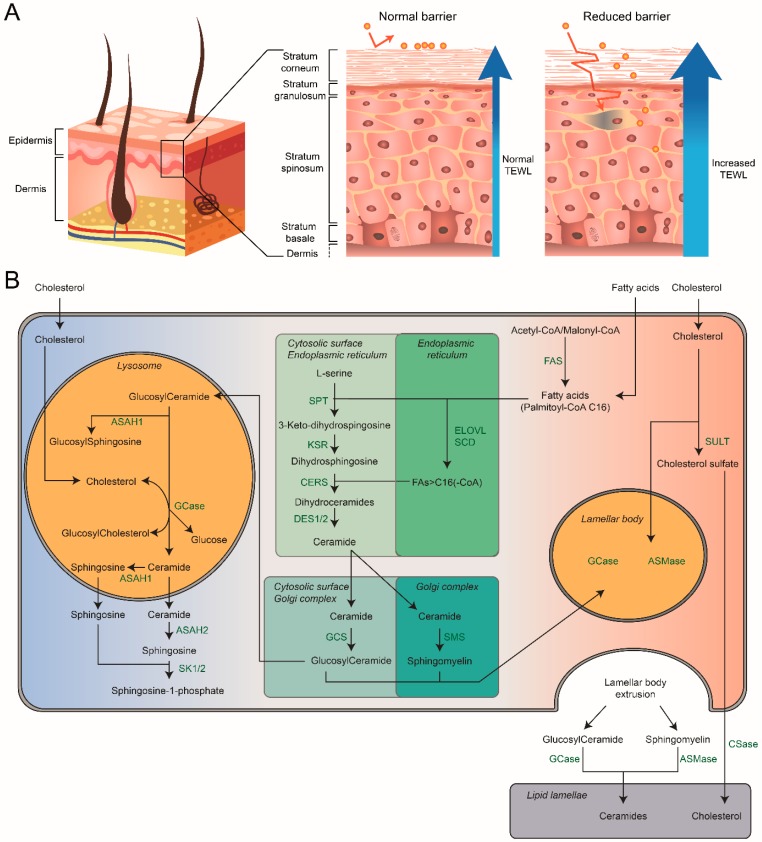
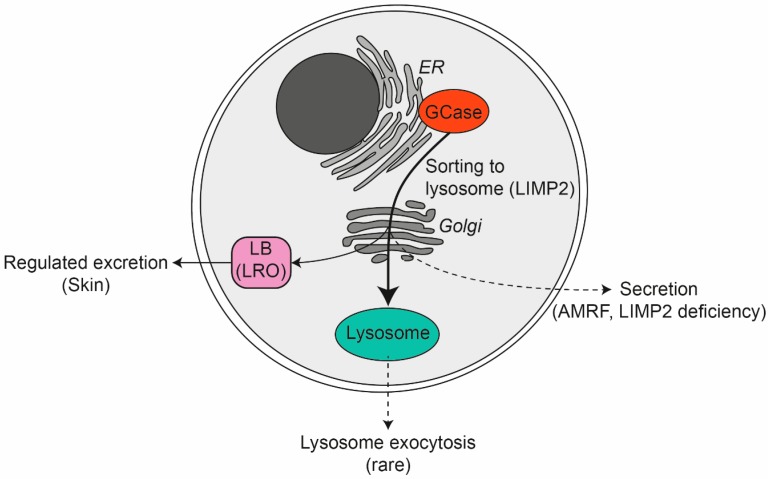
Glucocerebrosidase (GCase) is a retaining β-glucosidase with acid pH optimum metabolizing the glycosphingolipid glucosylceramide (GlcCer) to ceramide and glucose. Inherited deficiency of GCase causes the lysosomal storage disorder named Gaucher disease (GD). In GCase-deficient GD patients the accumulation of GlcCer in lysosomes of tissue macrophages is prominent. Based on the above, the key function of GCase as lysosomal hydrolase is well recognized, however it has become apparent that GCase fulfills in the human body at least one other key function beyond lysosomes. Crucially, GCase generates ceramides from GlcCer molecules in the outer part of the skin, a process essential for optimal skin barrier property and survival. This review covers the functions of GCase in and beyond lysosomes and also pays attention to the increasing insight in hitherto unexpected catalytic versatility of the enzyme.
Keywords: Gaucher disease; glucocerebrosidase; glucosylceramide; lysosome; skin.
Conflict of interest statement
The authors declare no conflicts of interest
Figures
References
-
- Gaucher P.C.E. Ph.D. Thesis. Faculté de Médecine; Paris, France: 1882. De L’epithelioma Primitif de la Rate, Hypertrophie Idiopathique de la Rate Sans Leucemie.
-
- Beutler E., Grabowski G.A. Glucosylceramide lipidosis-Gaucher disease. In: Sriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. McGraw-Hill; New York, NY, USA: 2001.
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
