

Variant effect predictions capture some aspects of deep mutational scanning experiments
- PMID: 32183714
- PMCID: PMC7077003
- DOI: 10.1186/s12859-020-3439-4
Variant effect predictions capture some aspects of deep mutational scanning experiments
Abstract
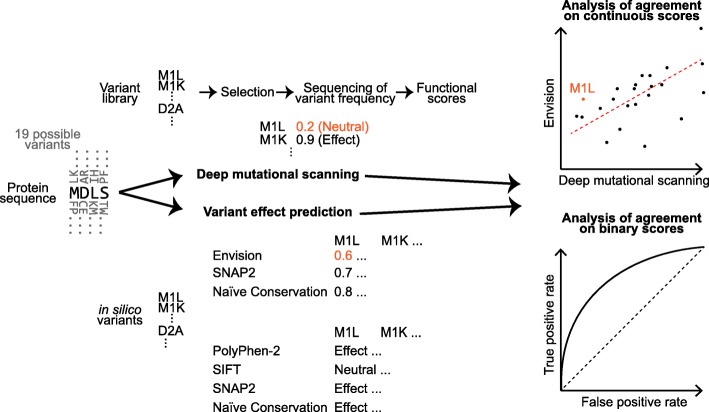
Background: Deep mutational scanning (DMS) studies exploit the mutational landscape of sequence variation by systematically and comprehensively assaying the effect of single amino acid variants (SAVs; also referred to as missense mutations, or non-synonymous Single Nucleotide Variants - missense SNVs or nsSNVs) for particular proteins. We assembled SAV annotations from 22 different DMS experiments and normalized the effect scores to evaluate variant effect prediction methods. Three trained on traditional variant effect data (PolyPhen-2, SIFT, SNAP2), a regression method optimized on DMS data (Envision), and a naïve prediction using conservation information from homologs.
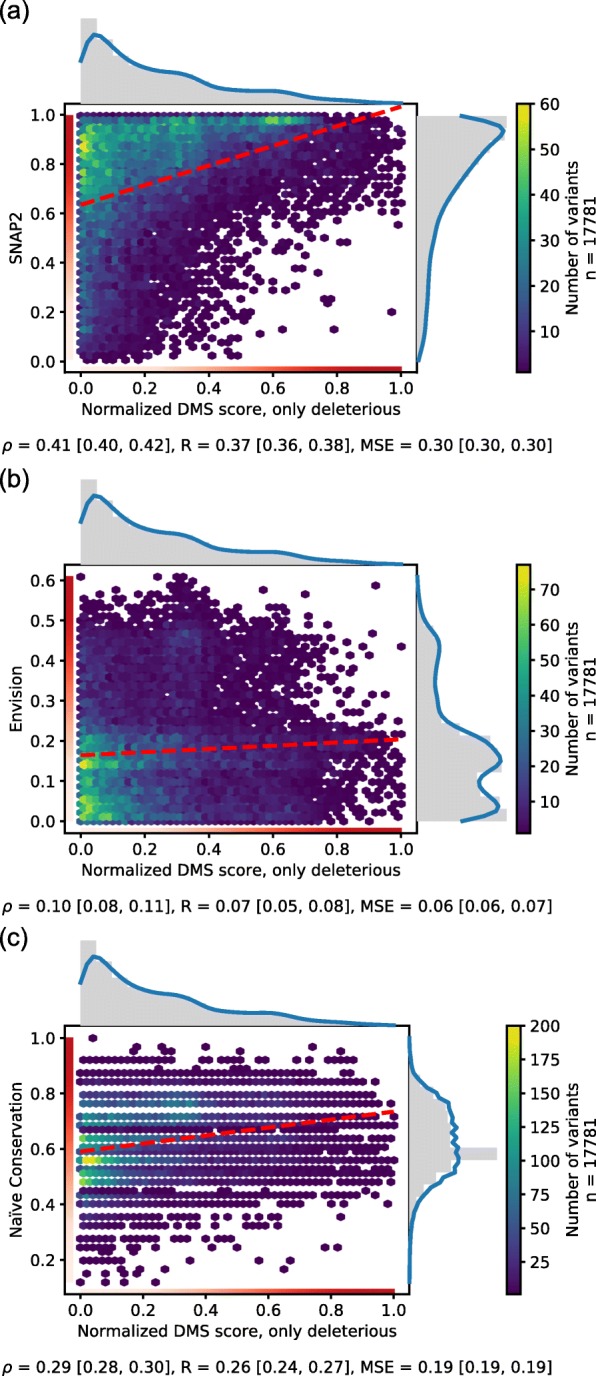
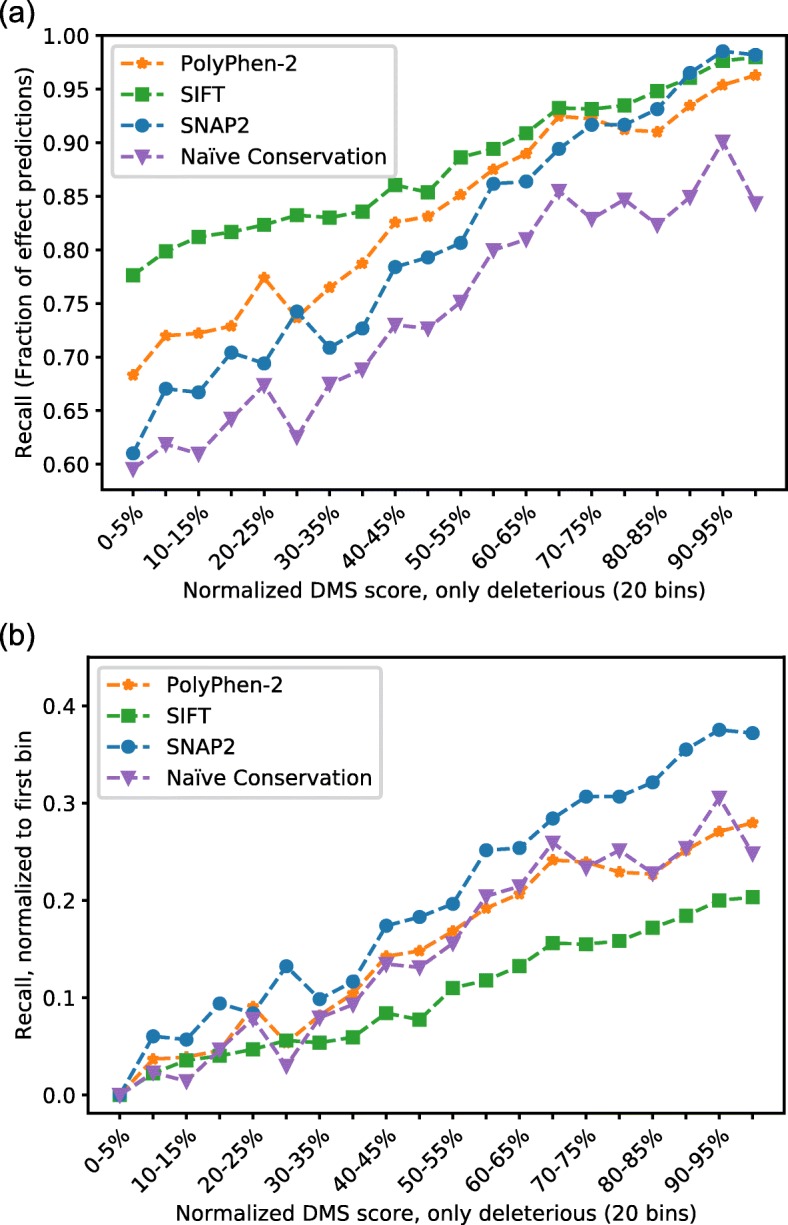
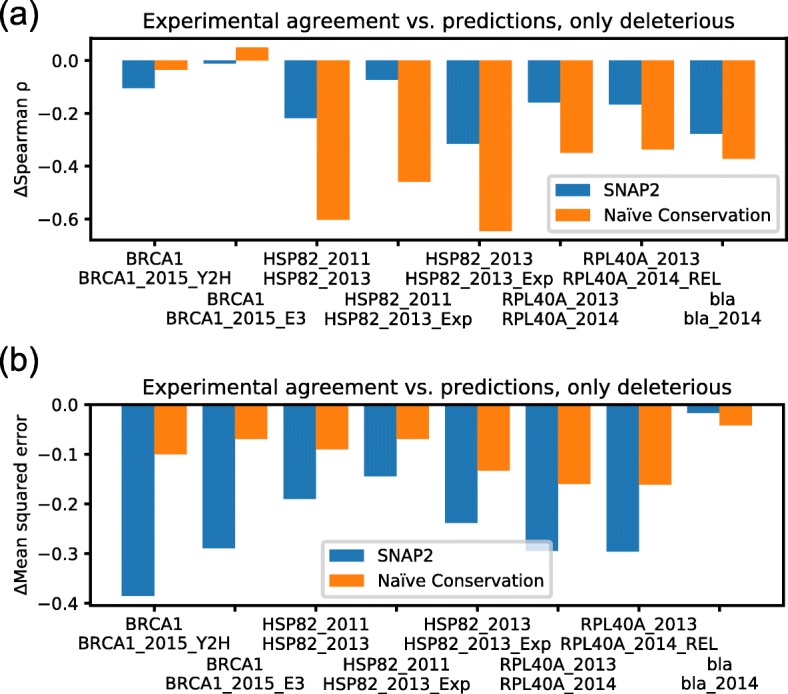
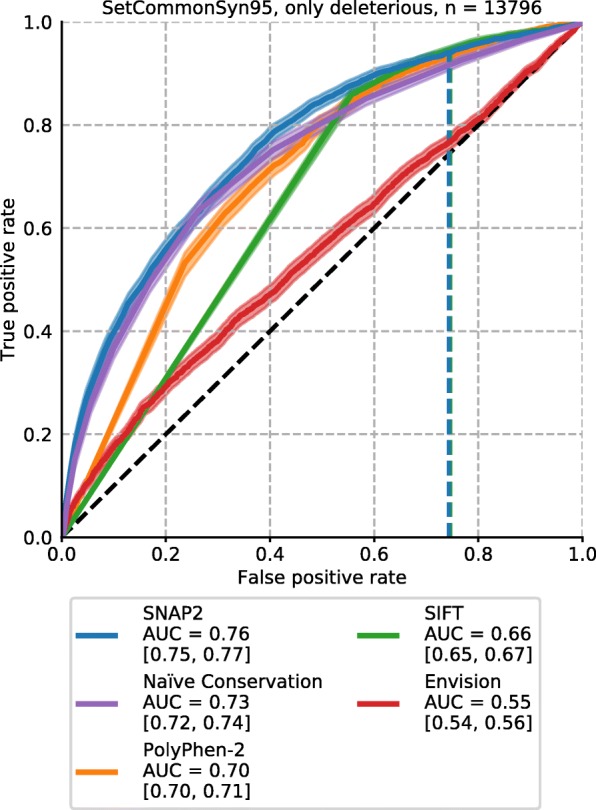
Results: On a set of 32,981 SAVs, all methods captured some aspects of the experimental effect scores, albeit not the same. Traditional methods such as SNAP2 correlated slightly more with measurements and better classified binary states (effect or neutral). Envision appeared to better estimate the precise degree of effect. Most surprising was that the simple naïve conservation approach using PSI-BLAST in many cases outperformed other methods. All methods captured beneficial effects (gain-of-function) significantly worse than deleterious (loss-of-function). For the few proteins with multiple independent experimental measurements, experiments differed substantially, but agreed more with each other than with predictions.
Conclusions: DMS provides a new powerful experimental means of understanding the dynamics of the protein sequence space. As always, promising new beginnings have to overcome challenges. While our results demonstrated that DMS will be crucial to improve variant effect prediction methods, data diversity hindered simplification and generalization.
Keywords: Deep mutational scanning; Missense variant; Non-synonymous sequence variant; Sequence variation; Single nucleotide variant; Variant effect prediction.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Embeddings from protein language models predict conservation and variant effects.Hum Genet. 2022 Oct;141(10):1629-1647. doi: 10.1007/s00439-021-02411-y. Epub 2021 Dec 30. Hum Genet. 2022. PMID: 34967936 Free PMC article.
-
Using deep mutational scanning to benchmark variant effect predictors and identify disease mutations.Mol Syst Biol. 2020 Jul;16(7):e9380. doi: 10.15252/msb.20199380. Mol Syst Biol. 2020. PMID: 32627955 Free PMC article.
-
Performance of in silico prediction tools for the classification of rare BRCA1/2 missense variants in clinical diagnostics.BMC Med Genomics. 2018 Mar 27;11(1):35. doi: 10.1186/s12920-018-0353-y. BMC Med Genomics. 2018. PMID: 29580235 Free PMC article.
-
News from the protein mutability landscape.J Mol Biol. 2013 Nov 1;425(21):3937-48. doi: 10.1016/j.jmb.2013.07.028. Epub 2013 Jul 26. J Mol Biol. 2013. PMID: 23896297 Review.
-
Objective assessment of the evolutionary action equation for the fitness effect of missense mutations across CAGI-blinded contests.Hum Mutat. 2017 Sep;38(9):1072-1084. doi: 10.1002/humu.23266. Epub 2017 Jun 21. Hum Mutat. 2017. PMID: 28544059 Free PMC article. Review.
Cited by
-
VariBench, new variation benchmark categories and data sets.Front Bioinform. 2023 Sep 19;3:1248732. doi: 10.3389/fbinf.2023.1248732. eCollection 2023. Front Bioinform. 2023. PMID: 37795169 Free PMC article. No abstract available.
-
Variant effect predictor correlation with functional assays is reflective of clinical classification performance.Genome Biol. 2025 Apr 22;26(1):104. doi: 10.1186/s13059-025-03575-w. Genome Biol. 2025. PMID: 40264194 Free PMC article.
-
Globally defining the effects of mutations in a picornavirus capsid.Elife. 2021 Jan 12;10:e64256. doi: 10.7554/eLife.64256. Elife. 2021. PMID: 33432927 Free PMC article.
-
Machine Learning-Guided Protein Engineering.ACS Catal. 2023 Oct 13;13(21):13863-13895. doi: 10.1021/acscatal.3c02743. eCollection 2023 Nov 3. ACS Catal. 2023. PMID: 37942269 Free PMC article. Review.
-
Embeddings from protein language models predict conservation and variant effects.Hum Genet. 2022 Oct;141(10):1629-1647. doi: 10.1007/s00439-021-02411-y. Epub 2021 Dec 30. Hum Genet. 2022. PMID: 34967936 Free PMC article.
References
-
- Tennessen Jacob A., Bigham Abigail W., O’Connor Timothy D., Fu Wenqing, Kenny Eimear E., Gravel Simon, McGee Sean, Do Ron, Liu Xiaoming, Jun Goo, Kang Hyun Min, Jordan Daniel, Leal Suzanne M., Gabriel Stacey, Rieder Mark J., Abecasis Goncalo, Altshuler David, Nickerson Deborah A., Boerwinkle Eric, Sunyaev Shamil, Bustamante Carlos D., Bamshad Michael J., Akey Joshua M. Evolution and Functional Impact of Rare Coding Variation from Deep Sequencing of Human Exomes. Science. 2012;337(6090):64–69. doi: 10.1126/science.1219240. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
