

Imaging Biomarkers for Neurodegeneration in Presymptomatic Familial Frontotemporal Lobar Degeneration
- PMID: 32184751
- PMCID: PMC7058699
- DOI: 10.3389/fneur.2020.00080
Imaging Biomarkers for Neurodegeneration in Presymptomatic Familial Frontotemporal Lobar Degeneration
Abstract
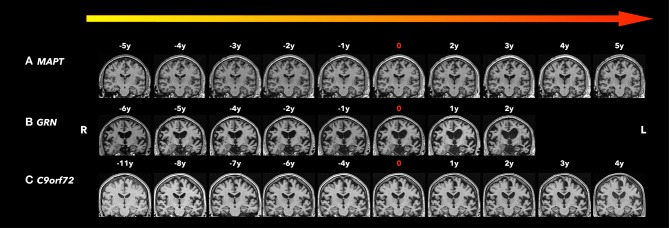
Frontotemporal lobar degeneration (FTLD) is a neurodegenerative disorder characterized by behavioral changes, language abnormality, as well as executive function deficits and motor impairment. In about 30-50% of FTLD patients, an autosomal dominant pattern of inheritance was found with major mutations in the MAPT, GRN, and the C9orf72 repeat expansion. These mutations could lead to neurodegenerative pathology years before clinical symptoms onset. With potential disease-modifying treatments that are under development, non-invasive biomarkers that help determine the early brain changes in presymptomatic FTLD patients will be critical for tracking disease progression and enrolling the right participants into the clinical trials at the right time during the disease course. In recent years, there is increasing evidence that a number of imaging biomarkers show the abnormalities during the presymptomatic stage. Imaging biomarkers of presymptomatic familial FTLD may provide insight into the underlying neurodegenerative process years before symptom onset. Structural magnetic resonance imaging (MRI) has demonstrated cortical degeneration with a mutation-specific neurodegeneration pattern years before onset of clinical symptoms in presymptomatic familial FTLD mutation carriers. In addition, diffusion tensor imaging (DTI) has shown the loss of white matter microstructural integrity in the presymptomatic stage of familial FTLD. Furthermore, proton magnetic resonance spectroscopy (1H MRS), which provides a non-invasive measurement of brain biochemistry, has identified early neurochemical abnormalities in presymptomatic MAPT mutation carriers. Positron emission tomography (PET) imaging with [18F]-fluorodeoxyglucose (FDG) has demonstrated the glucose hypometabolism in the presymptomatic stage of familial FTLD. Also, a novel PET ligand, 18F-AV-1451, has been used in this group to evaluate tau deposition in the brain. Promising imaging biomarkers for presymptomatic familial FTLD have been identified and assessed for specificity and sensitivity for accurate prediction of symptom onset and tracking disease progression during the presymptomatic stage when clinical measures are not useful. Furthermore, identifying imaging biomarkers for the presymptomatic stage is important for the design of disease-modifying trials. We review the recent progress in imaging biomarkers of the presymptomatic phase of familial FTLD and discuss the imaging techniques and analysis methods, with a focus on the potential implication of these imaging techniques and their utility in specific mutation types.
Keywords: C9orf72; GRN; MAPT; familial frontotemporal lobar degeneration; imaging biomarker; presymptomatic.
Copyright © 2020 Chen and Kantarci.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous