

Hyperinsulinemic hypoglycemia in children and adolescents: Recent advances in understanding of pathophysiology and management
- PMID: 32185602
- PMCID: PMC7560934
- DOI: 10.1007/s11154-020-09548-7
Hyperinsulinemic hypoglycemia in children and adolescents: Recent advances in understanding of pathophysiology and management
Abstract
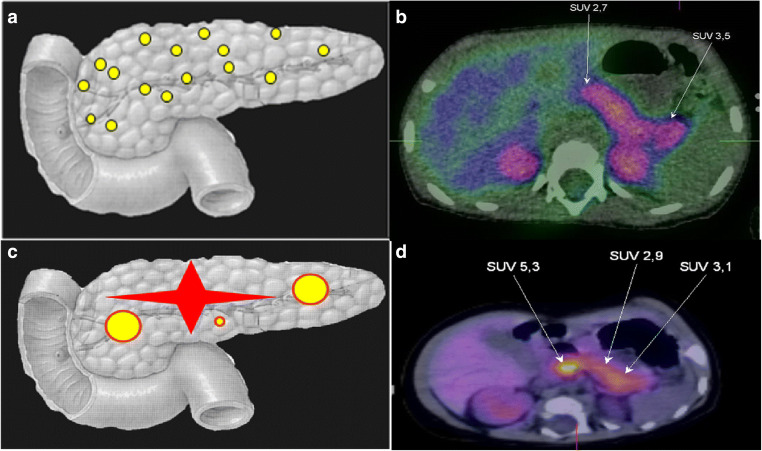
Hyperinsulinemic hypoglycemia (HH) is characterized by unregulated insulin release, leading to persistently low blood glucose concentrations with lack of alternative fuels, which increases the risk of neurological damage in these patients. It is the most common cause of persistent and recurrent hypoglycemia in the neonatal period. HH may be primary, Congenital HH (CHH), when it is associated with variants in a number of genes implicated in pancreatic development and function. Alterations in fifteen genes have been recognized to date, being some of the most recently identified mutations in genes HK1, PGM1, PMM2, CACNA1D, FOXA2 and EIF2S3. Alternatively, HH can be secondary when associated with syndromes, intra-uterine growth restriction, maternal diabetes, birth asphyxia, following gastrointestinal surgery, amongst other causes. CHH can be histologically characterized into three groups: diffuse, focal or atypical. Diffuse and focal forms can be determined by scanning using fluorine-18 dihydroxyphenylalanine-positron emission tomography. Newer and improved isotopes are currently in development to provide increased diagnostic accuracy in identifying lesions and performing successful surgical resection with the ultimate aim of curing the condition. Rapid diagnostics and innovative methods of management, including a wider range of treatment options, have resulted in a reduction in co-morbidities associated with HH with improved quality of life and long-term outcomes. Potential future developments in the management of this condition as well as pathways to transition of the care of these highly vulnerable children into adulthood will also be discussed.
Keywords: 18F-DOPA-PET; Hyperinsulinism; Hypoglycemia; Lanreotide; Sirolimus; Transition to adult services.
Conflict of interest statement
Nick Oliver has received honoraria for advisory board participation or speaking from Dexcom, Roche diabetes, and Medtronic diabetes; support for education from Novo Nordisk and Takeda; and research funding from Dexcom and Roche diabetes. The other authors have no information to disclose.
Figures


References
-
- Ahrén B. Autonomic regulation of islet hormone secretion--implications for health and disease. Diabetologia. 2000;43(4):393–410. - PubMed
-
- Senniappan S, Shanti B, James C, Hussain K. Hyperinsulinaemic hypoglycaemia: genetic mechanisms, diagnosis and management. J Inherit Metab Dis. 2012;35(4):589–601. - PubMed
-
- Chinoy, A. et al. ‘Focal congenital hyperinsulinism as a cause for sudden infant death’. Pediatr Dev Pathol. 2019;22(1):65–69. - PubMed
-
- Guyot A, Moreau F, Eberhard M, Gaulier JM, Paraf F. Congenital hyperinsulinism revealed by sudden infant death. Ann Pathol. 2017;37(5):429–432. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
