

Defining Epidermal Basal Cell States during Skin Homeostasis and Wound Healing Using Single-Cell Transcriptomics
- PMID: 32187560
- PMCID: PMC7218802
- DOI: 10.1016/j.celrep.2020.02.091
Defining Epidermal Basal Cell States during Skin Homeostasis and Wound Healing Using Single-Cell Transcriptomics
Abstract
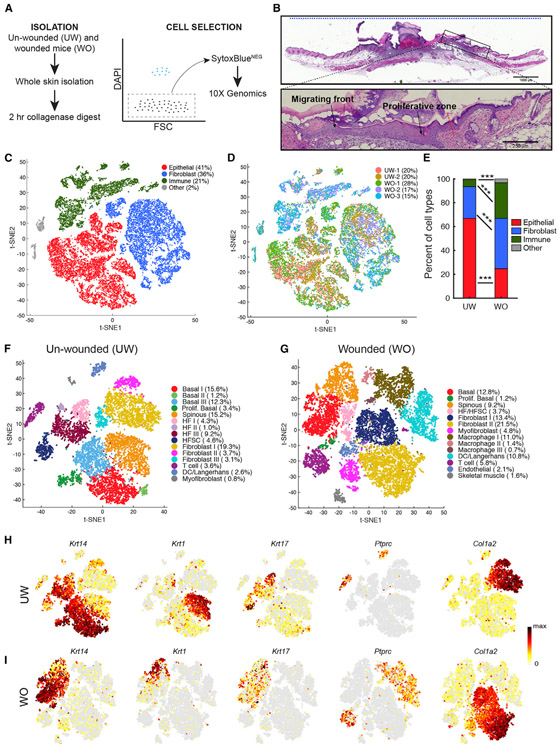
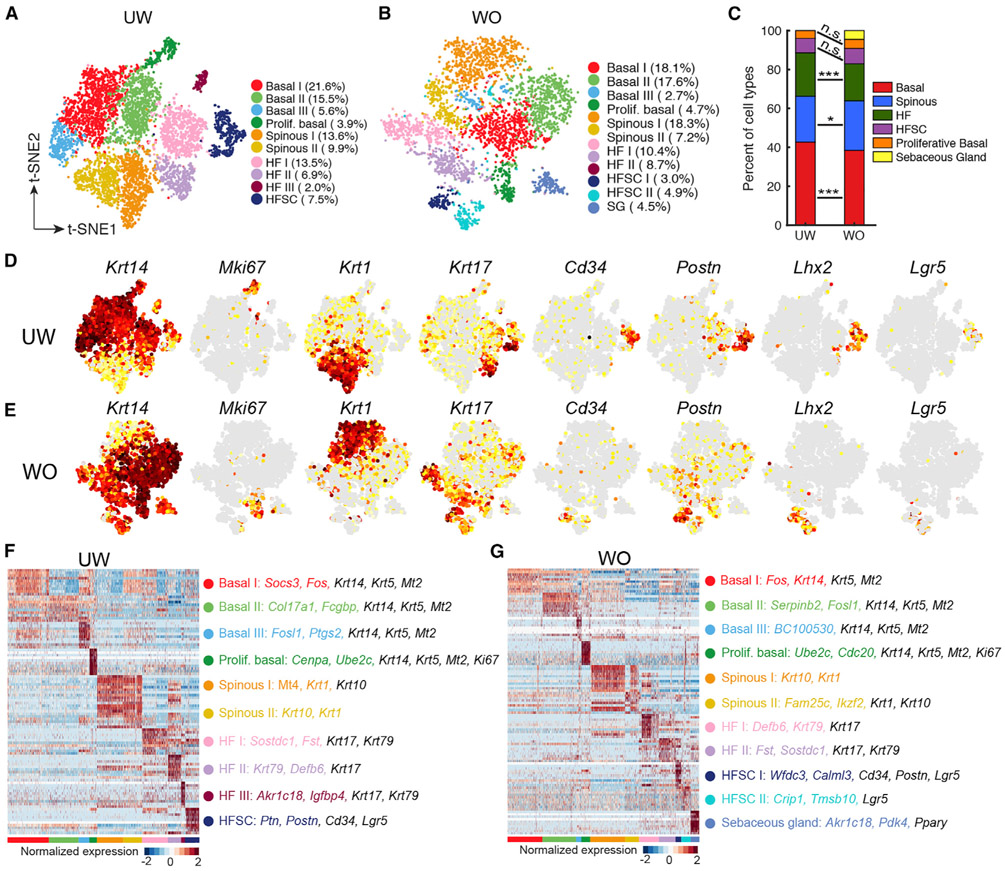
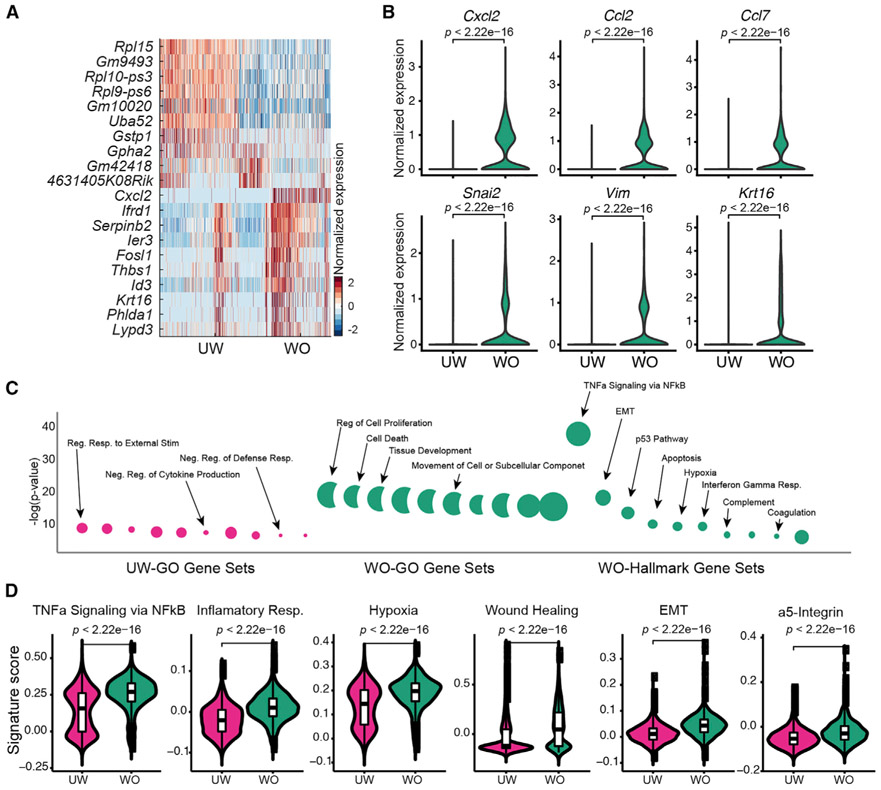
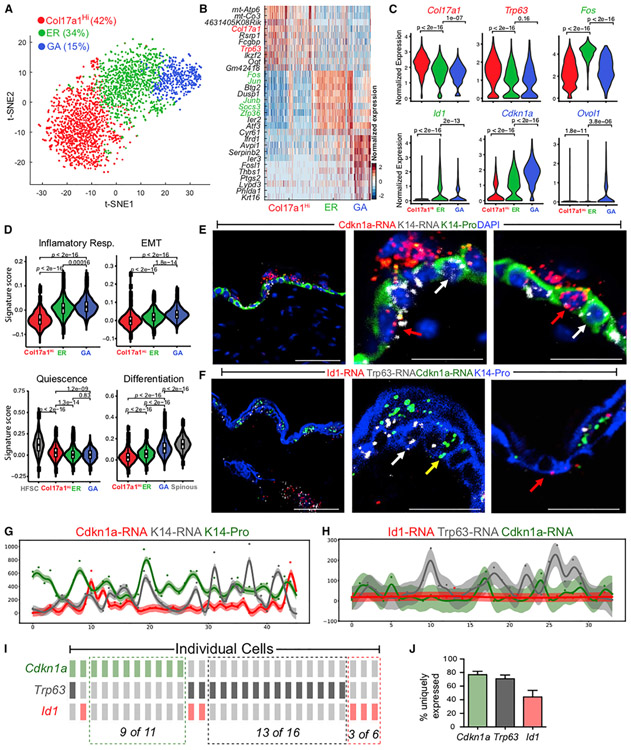
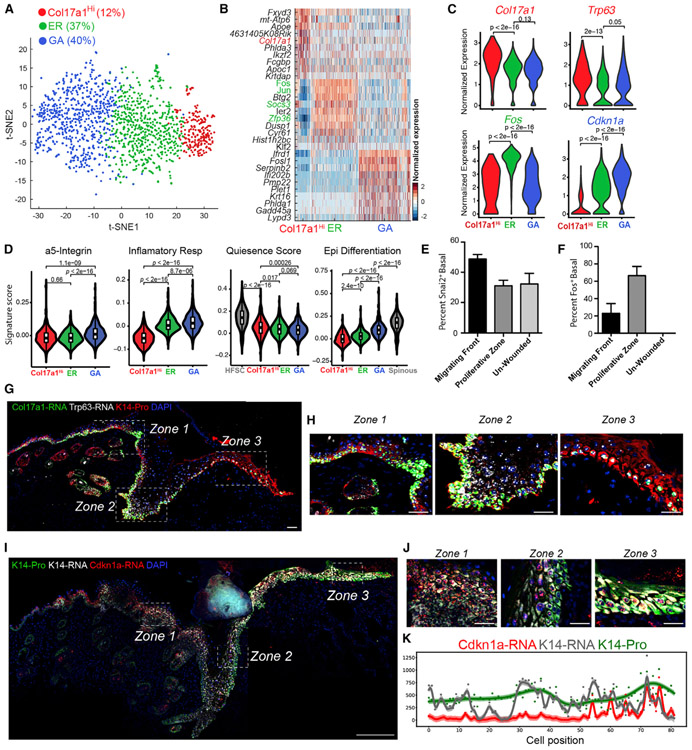
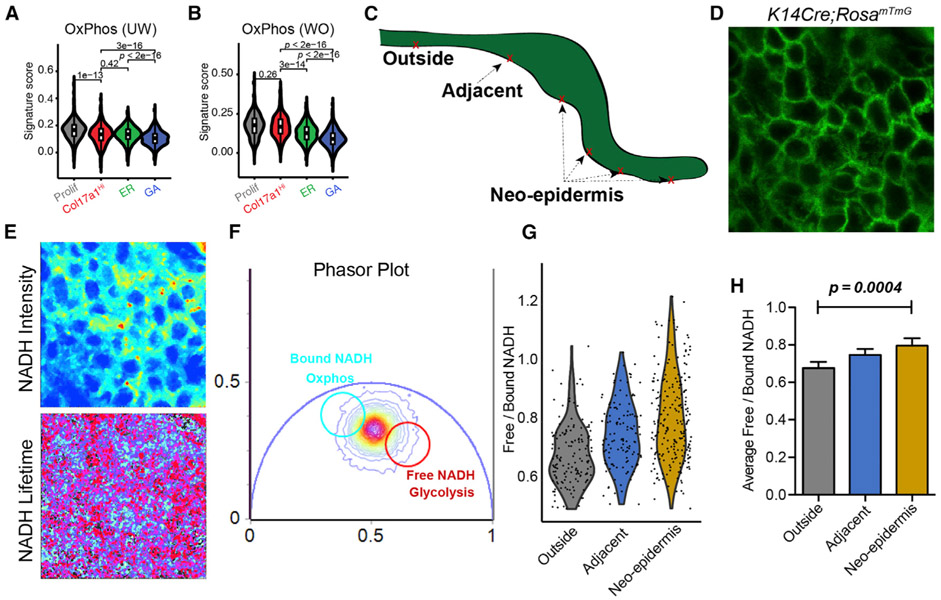
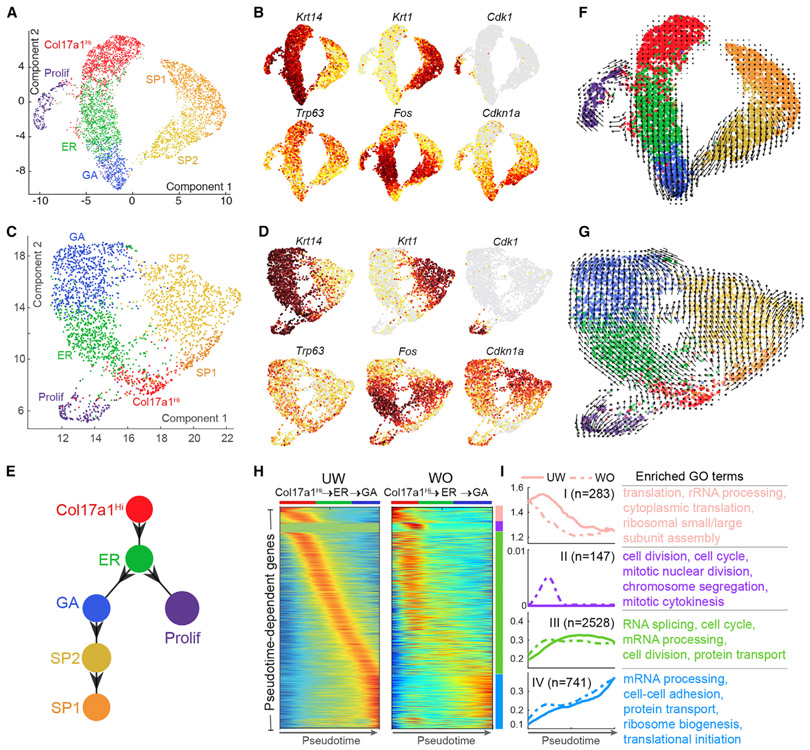
Our knowledge of transcriptional heterogeneities in epithelial stem and progenitor cell compartments is limited. Epidermal basal cells sustain cutaneous tissue maintenance and drive wound healing. Previous studies have probed basal cell heterogeneity in stem and progenitor potential, but a comprehensive dissection of basal cell dynamics during differentiation is lacking. Using single-cell RNA sequencing coupled with RNAScope and fluorescence lifetime imaging, we identify three non-proliferative and one proliferative basal cell state in homeostatic skin that differ in metabolic preference and become spatially partitioned during wound re-epithelialization. Pseudotemporal trajectory and RNA velocity analyses predict a quasi-linear differentiation hierarchy where basal cells progress from Col17a1Hi/Trp63Hi state to early-response state, proliferate at the juncture of these two states, or become growth arrested before differentiating into spinous cells. Wound healing induces plasticity manifested by dynamic basal-spinous interconversions at multiple basal transcriptional states. Our study provides a systematic view of epidermal cellular dynamics, supporting a revised "hierarchical-lineage" model of homeostasis.
Keywords: FLIM; basal cell state; cellular transition dynamics; epidermis; metabolism; plasticity; single-cell RNA sequencing; skin; stem cell; wound healing.
Copyright © 2020 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
References
-
- Andl T, Ahn K, Kairo A, Chu EY, Wine-Lee L, Reddy ST, Croft NJ, Cebra-Thomas JA, Metzger D, Chambon P, et al. (2004). Epithelial Bmpr1a regulates differentiation and proliferation in postnatal hair follicles and is essential for tooth development. Development 131, 2257–2268. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
