

Inheritance of mitochondrial DNA in humans: implications for rare and common diseases
- PMID: 32187761
- PMCID: PMC8641369
- DOI: 10.1111/joim.13047
Inheritance of mitochondrial DNA in humans: implications for rare and common diseases
Abstract
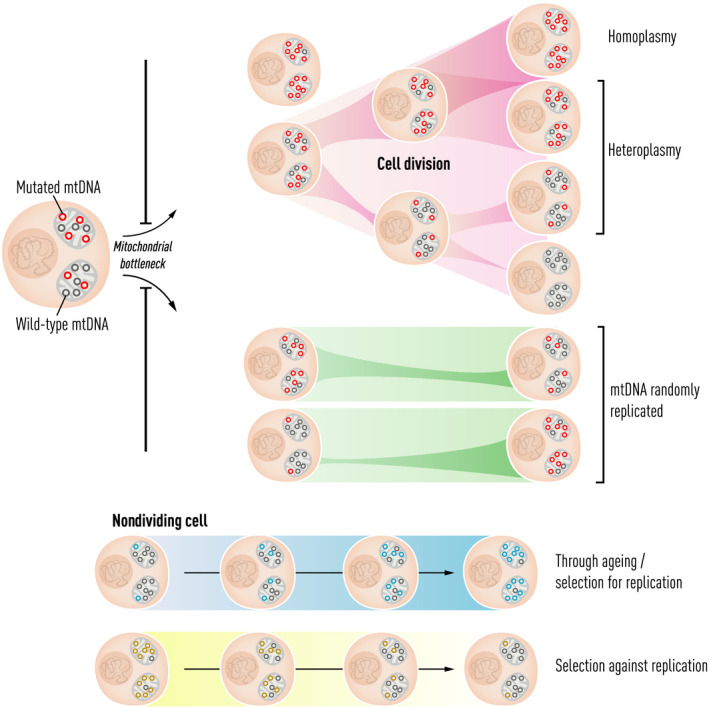
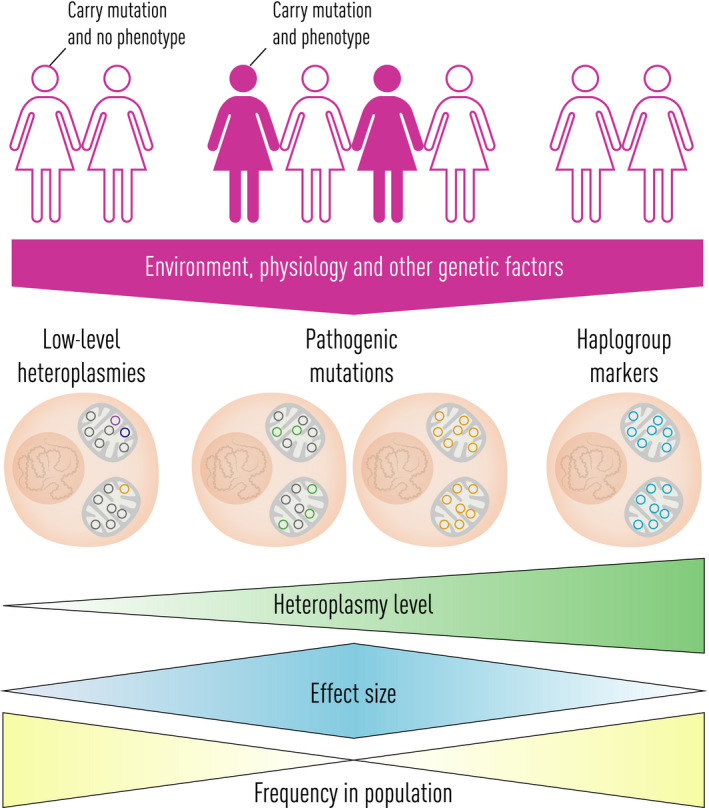
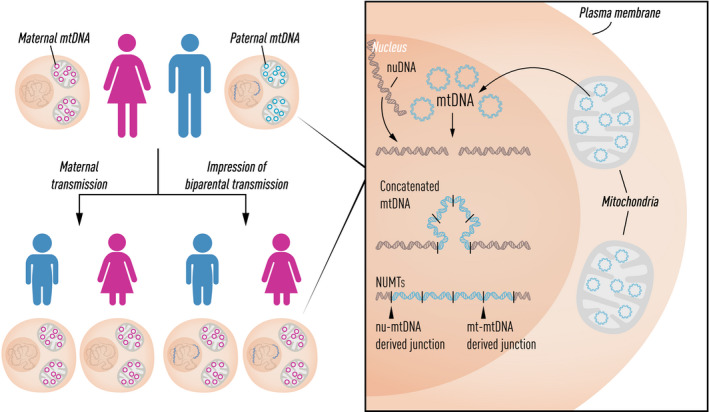
The first draft human mitochondrial DNA (mtDNA) sequence was published in 1981, paving the way for two decades of discovery linking mtDNA variation with human disease. Severe pathogenic mutations cause sporadic and inherited rare disorders that often involve the nervous system. However, some mutations cause mild organ-specific phenotypes that have a reduced clinical penetrance, and polymorphic variation of mtDNA is associated with an altered risk of developing several late-onset common human diseases including Parkinson's disease. mtDNA mutations also accumulate during human life and are enriched in affected organs in a number of age-related diseases. Thus, mtDNA contributes to a wide range of human pathologies. For many decades, it has generally been accepted that mtDNA is inherited exclusively down the maternal line in humans. Although recent evidence has challenged this dogma, whole-genome sequencing has identified nuclear-encoded mitochondrial sequences (NUMTs) that can give the false impression of paternally inherited mtDNA. This provides a more likely explanation for recent reports of 'bi-parental inheritance', where the paternal alleles are actually transmitted through the nuclear genome. The presence of both mutated and wild-type variant alleles within the same individual (heteroplasmy) and rapid shifts in allele frequency can lead to offspring with variable severity of disease. In addition, there is emerging evidence that selection can act for and against specific mtDNA variants within the developing germ line, and possibly within developing tissues. Thus, understanding how mtDNA is inherited has far-reaching implications across medicine. There is emerging evidence that this highly dynamic system is amenable to therapeutic manipulation, raising the possibility that we can harness new understanding to prevent and treat rare and common human diseases where mtDNA mutations play a key role.
Keywords: human mitochondrial DNA; mitochondrial DNA mutation; mitochondrial bottleneck; mitochondrial disorders; mitochondrial inheritance.
© 2020 The Authors. Journal of Internal Medicine published by John Wiley & Sons Ltd on behalf of Association for Publication of The Journal of Internal Medicine.
Conflict of interest statement
No conflicts of interest were declared.
Figures
References
-
- Wallace DC. Mitochondrial genetic medicine. Nat Genet 2018; 50: 1642–9. - PubMed
-
- Stewart JB, Chinnery PF. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet 2015; 16: 530–42. - PubMed
-
- Vafai SB, Mootha VK. Medicine. A common pathway for a rare disease? Science 2013; 342: 1453–4. - PubMed
-
- Wallace DC, Singh G, Lott MT et al. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science 1988; 242: 1427–30. - PubMed
-
- Holt I, Harding AE, Morgan‐Hughes JA. Deletion of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988; 331: 717–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
