

Affinity-Bead Assisted Mass Spectrometry (Affi-BAMS): A Multiplexed Microarray Platform for Targeted Proteomics
- PMID: 32188029
- PMCID: PMC7139916
- DOI: 10.3390/ijms21062016
Affinity-Bead Assisted Mass Spectrometry (Affi-BAMS): A Multiplexed Microarray Platform for Targeted Proteomics
Abstract
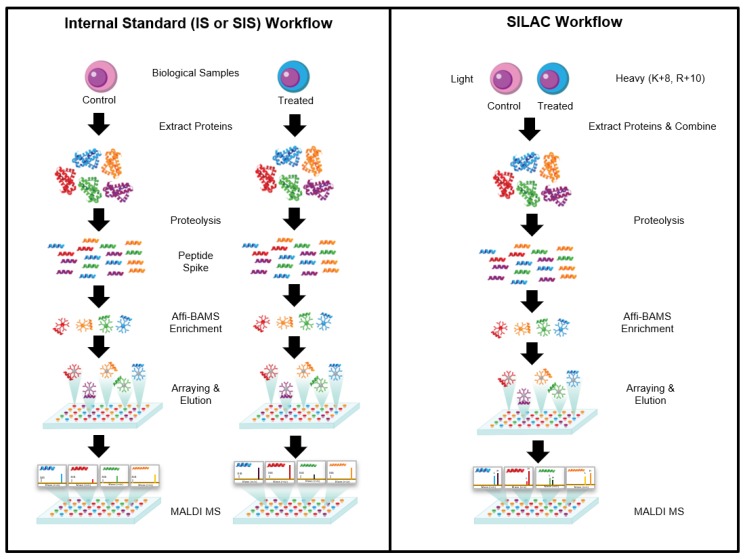
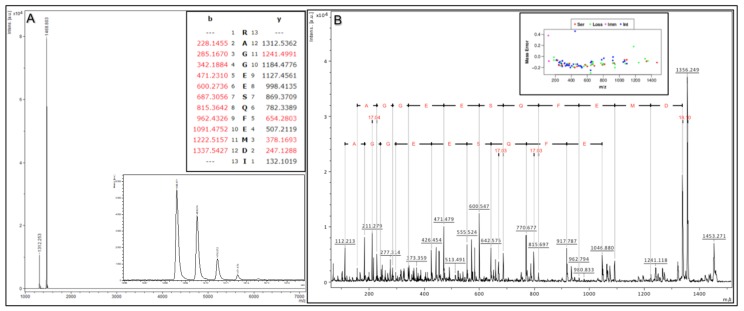
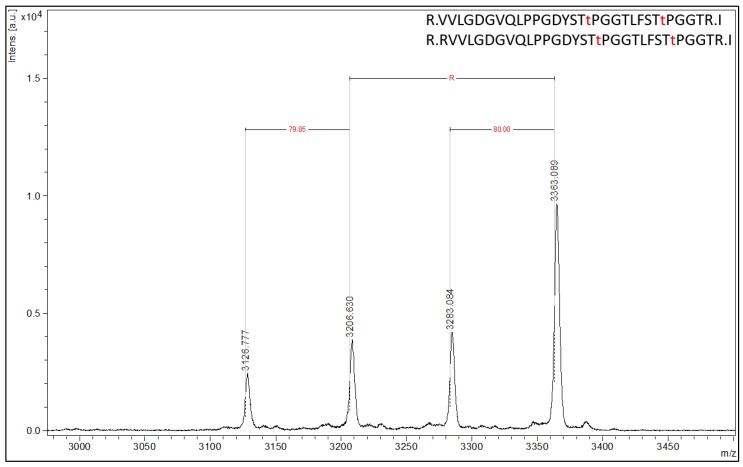
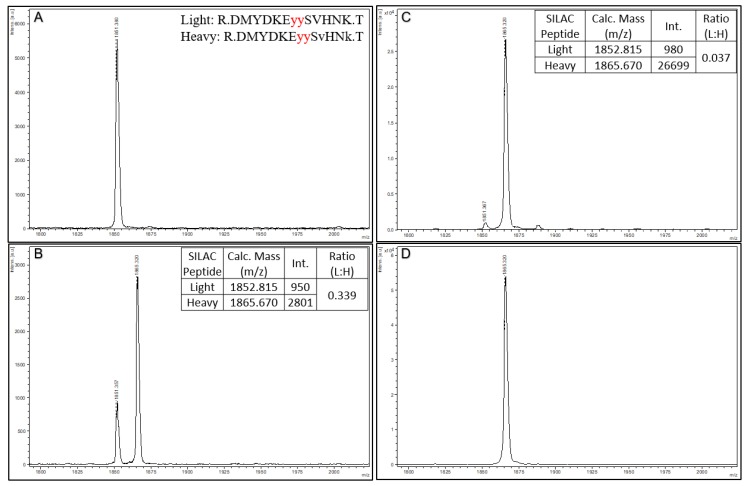
The ability to quantitatively probe diverse panels of proteins and their post-translational modifications (PTMs) across multiple samples would aid a broad spectrum of biological, biochemical and pharmacological studies. We report a novel, microarray analytical technology that combines immuno-affinity capture with Matrix Assisted Laser Desorption Ionization Mass Spectrometry (MALDI MS), which is capable of supporting highly multiplexed, targeted proteomic assays. Termed "Affinity-Bead Assisted Mass Spectrometry" (Affi-BAMS), this LC-free technology enables development of highly specific and customizable assay panels for simultaneous profiling of multiple proteins and PTMs. While affinity beads have been used previously in combination with MS, the Affi-BAMS workflow uses enrichment on a single bead that contains one type of antibody, generally capturing a single analyte (protein or PTM) while having enough binding capacity to enable quantification within approximately 3 orders of magnitude. The multiplexing capability is achieved by combining Affi-BAMS beads with different protein specificities. To enable screening of bead-captured analytes by MS, we further developed a novel method of performing spatially localized elution of targets from individual beads arrayed on a microscope slide. The resulting arrays of micro spots contain highly concentrated analytes localized within 0.5 mm diameter spots that can be directly measured using MALDI MS. While both intact proteins and protein fragments can be monitored by Affi-BAMS, we initially focused on applying this technology for bottom-up proteomics to enable screening of hundreds of samples per day by combining the robust magnetic bead-based workflow with the high throughput nature of MALDI MS acquisition. To demonstrate the variety of applications and robustness of Affi-BAMS, several studies are presented that focus on the response of 4EBP1, RPS6, ERK1/ERK2, mTOR, Histone H3 and C-MET to stimuli including rapamycin, H2O2, EPO, SU11274, Staurosporine and Vorinostat.
Keywords: BAMS; MALDI MS; PTMs; bead assisted mass spectrometry; multiplex assays; targeted proteomics.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures

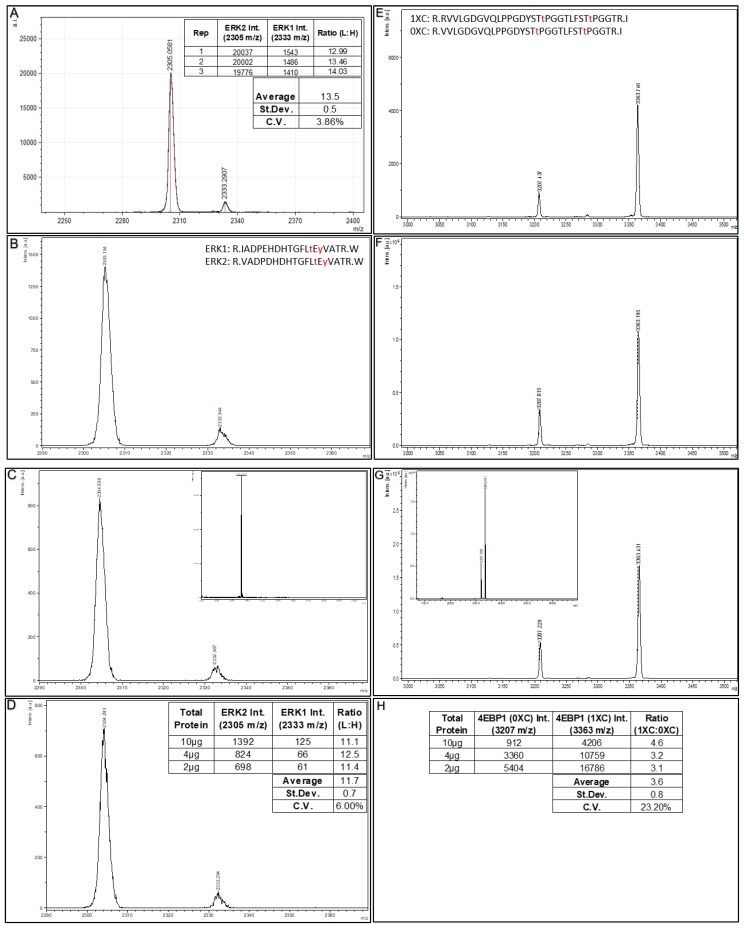
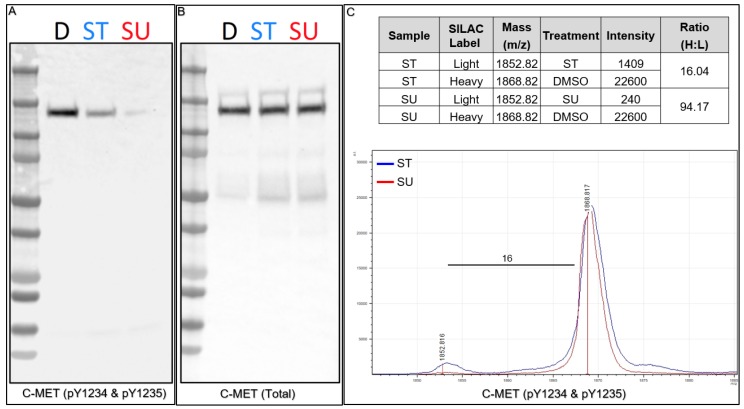
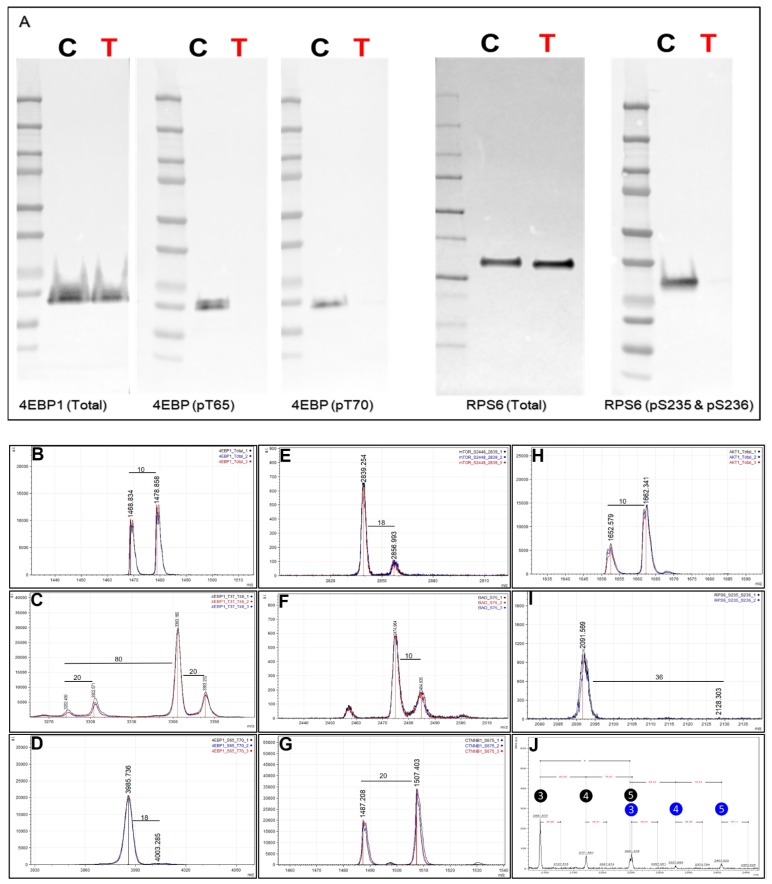
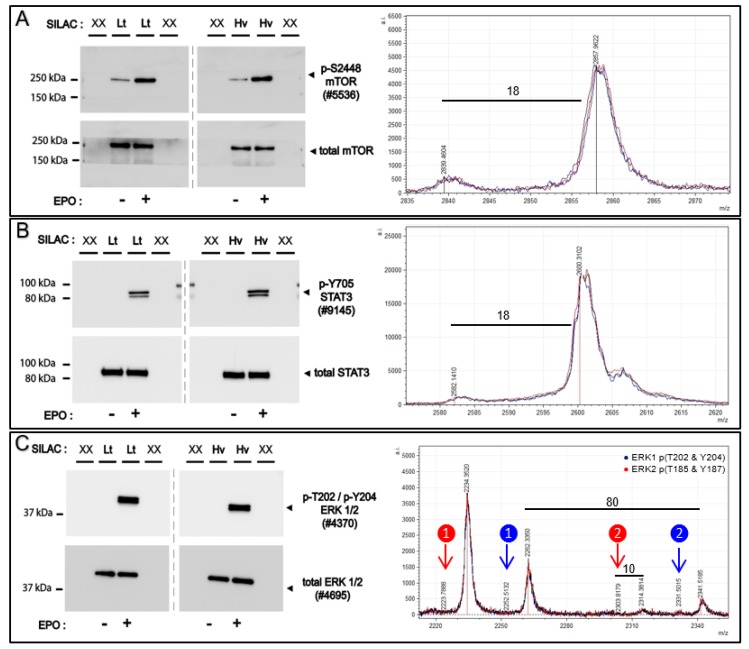

References
-
- Meier F., Brunner A.-D., Koch S., Koch H., Lubeck M., Krause M., Goedecke N., Decker J., Kosinski T., Park M.A., et al. Online Parallel Accumulation–Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer. Mol. Cell. Proteom. 2018;17:2534–2545. doi: 10.1074/mcp.TIR118.000900. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
