

Time course regulatory analysis based on paired expression and chromatin accessibility data
- PMID: 32188700
- PMCID: PMC7197475
- DOI: 10.1101/gr.257063.119
Time course regulatory analysis based on paired expression and chromatin accessibility data
Abstract
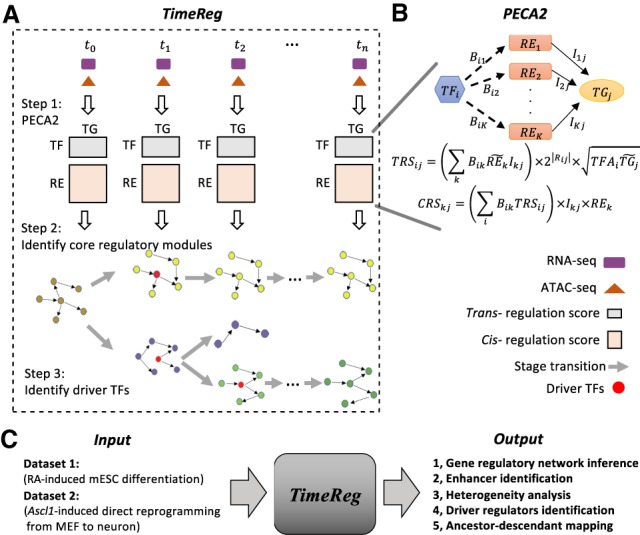
A time course experiment is a widely used design in the study of cellular processes such as differentiation or response to stimuli. In this paper, we propose
© 2020 Duren et al.; Published by Cold Spring Harbor Laboratory Press.
Figures








References
-
- Chen J, Kubalak SW, Chien KR. 1998. Ventricular muscle-restricted targeting of the RXRα gene reveals a non-cell-autonomous requirement in cardiac chamber morphogenesis. Development 125: 1943–1949. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials