

Epidermal Growth Factor Receptor and Abl2 Kinase Regulate Distinct Steps of Human Papillomavirus 16 Endocytosis
- PMID: 32188731
- PMCID: PMC7269448
- DOI: 10.1128/JVI.02143-19
Epidermal Growth Factor Receptor and Abl2 Kinase Regulate Distinct Steps of Human Papillomavirus 16 Endocytosis
Abstract
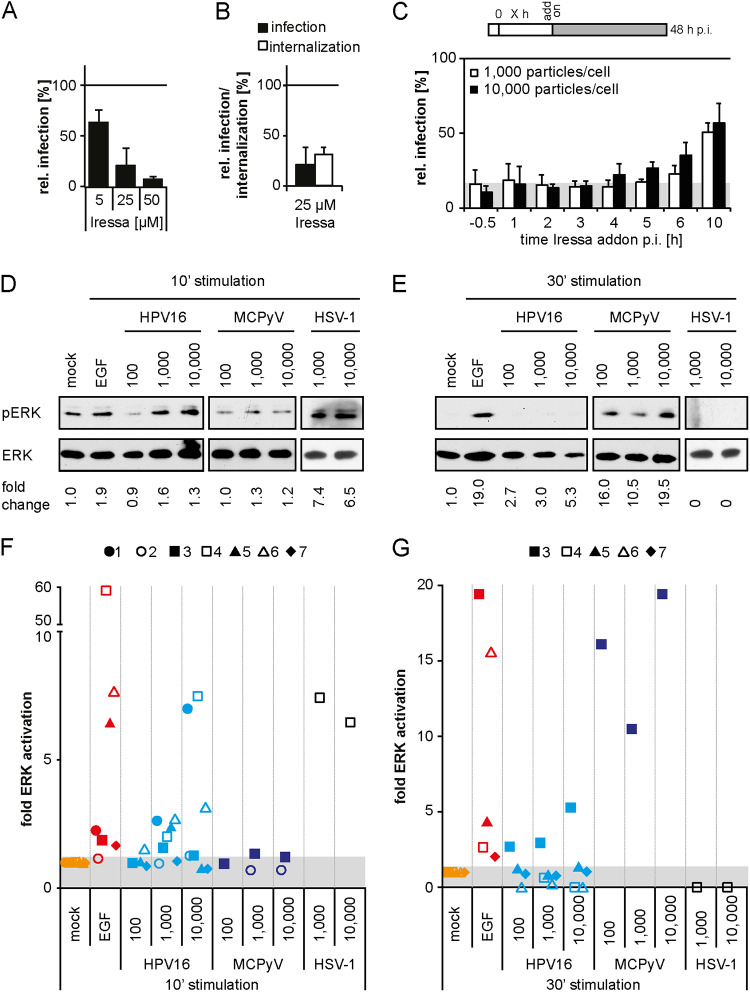
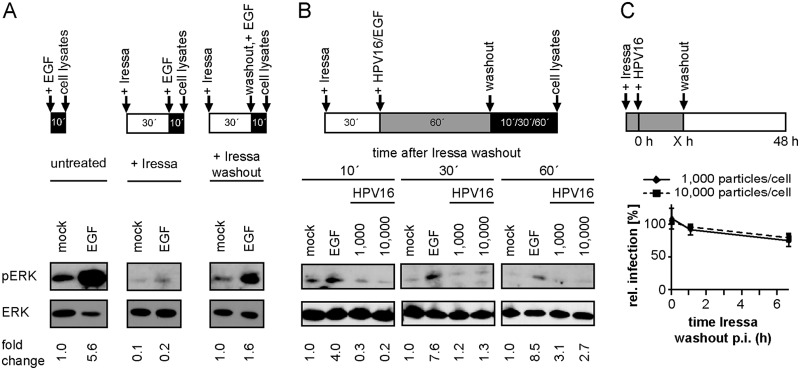
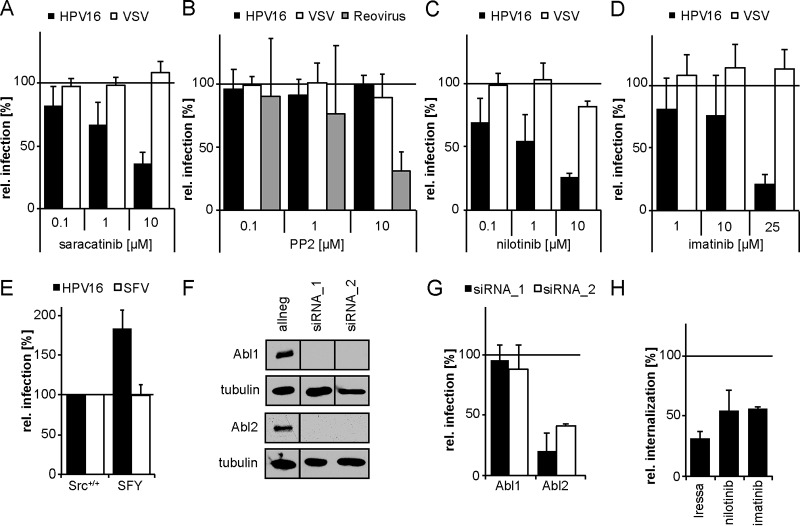
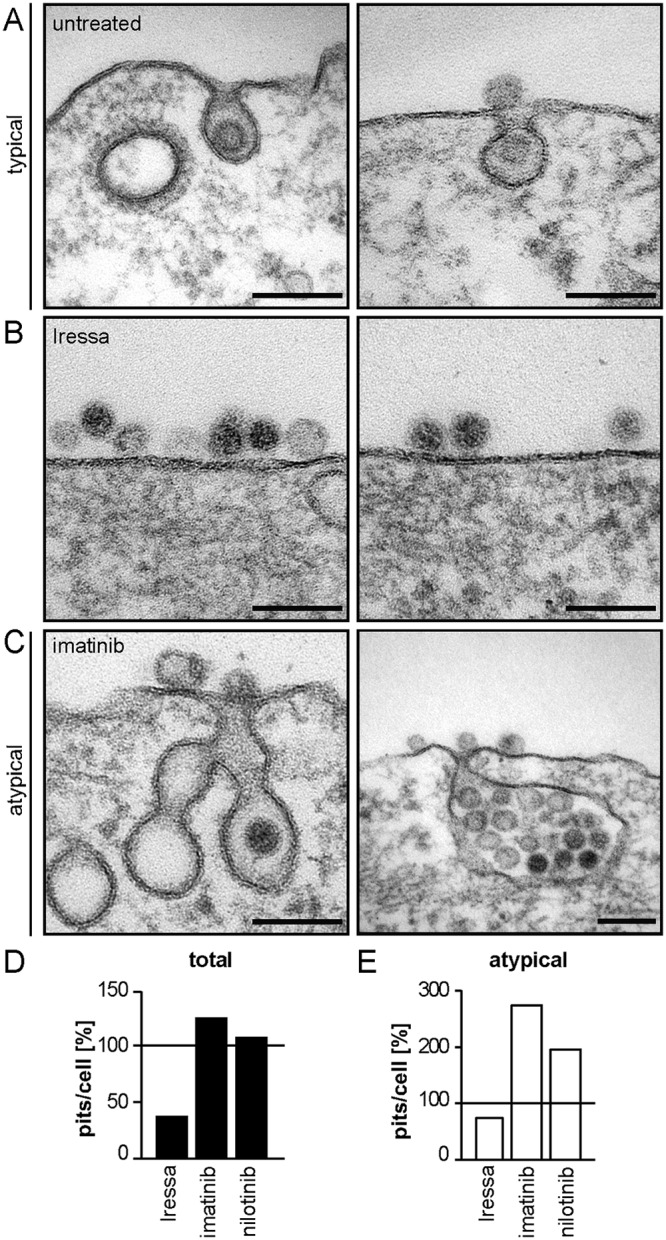
Human papillomavirus 16 (HPV16), the leading cause of cervical cancer, exploits a novel endocytic pathway during host cell entry. This mechanism shares many requirements with macropinocytosis but differs in the mode of vesicle formation. Previous work indicated a role of the epidermal growth factor receptor (EGFR) in HPV16 endocytosis. However, the functional outcome of EGFR signaling and its downstream targets during HPV16 uptake are not well characterized. Here, we analyzed the functional importance of signal transduction via EGFR and its downstream effectors for endocytosis of HPV16. Our findings indicate two phases of EGFR signaling as follows: a-likely dispensable-transient activation with or shortly after cell binding and signaling required throughout the process of asynchronous internalization of HPV16. Interestingly, EGFR inhibition interfered with virus internalization and strongly reduced the number of endocytic pits, suggesting a role for EGFR signaling in the induction of HPV16 endocytosis. Moreover, we identified the Src-related kinase Abl2 as a novel regulator of virus uptake. Inhibition of Abl2 resulted in an accumulation of misshaped endocytic pits, indicating Abl2's importance for endocytic vesicle maturation. Since Abl2 rather than Src, a regulator of membrane ruffling during macropinocytosis, mediated downstream signaling of EGFR, we propose that the selective effector targeting downstream of EGFR determines whether HPV16 endocytosis or macropinocytosis is induced.IMPORTANCE Human papillomaviruses are small, nonenveloped DNA viruses that infect skin and mucosa. The so-called high-risk HPVs (e.g., HPV16, HPV18, HPV31) have transforming potential and are associated with various anogenital and oropharyngeal tumors. These viruses enter host cells by a novel endocytic pathway with unknown cellular function. To date, it is unclear how endocytic vesicle formation occurs mechanistically. Here, we addressed the role of epidermal growth factor receptor signaling, which has previously been implicated in HPV16 endocytosis and identified the kinase Abl2 as a novel regulator of virus uptake. Since other viruses, such as influenza A virus and lymphocytic choriomeningitis virus, possibly make use of related mechanisms, our findings shed light on fundamental strategies of virus entry and may in turn help to develop new host cell-targeted antiviral strategies.
Keywords: HPV; endocytosis; papillomavirus; signaling; virus entry.
Copyright © 2020 American Society for Microbiology.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
