Environmental remodeling of human gut microbiota and antibiotic resistome in livestock farms
- PMID: 32188862
- PMCID: PMC7080799
- DOI: 10.1038/s41467-020-15222-y
Environmental remodeling of human gut microbiota and antibiotic resistome in livestock farms
Abstract
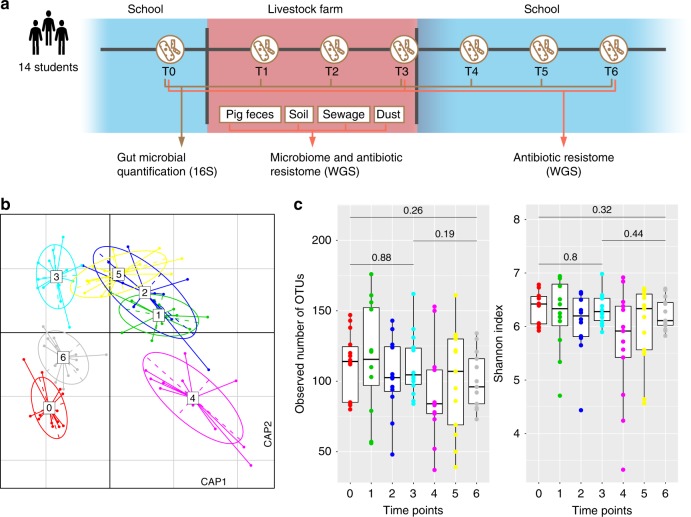
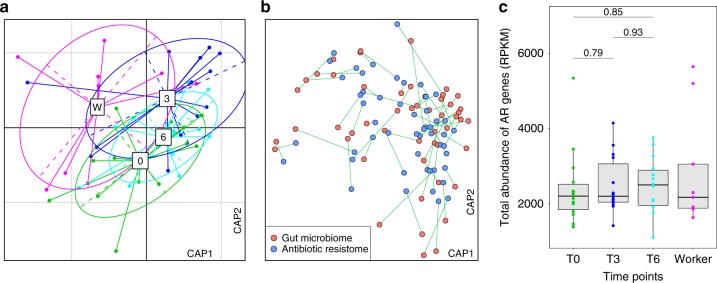
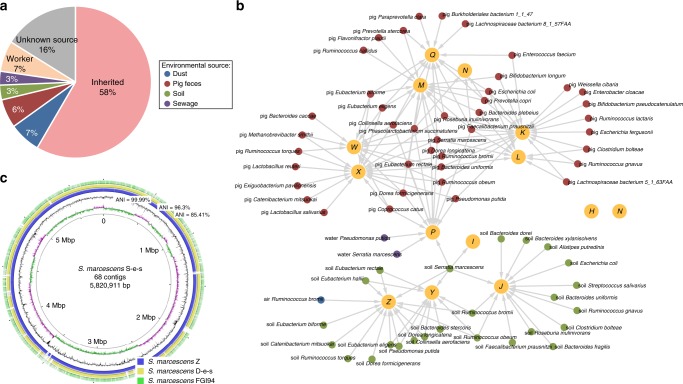
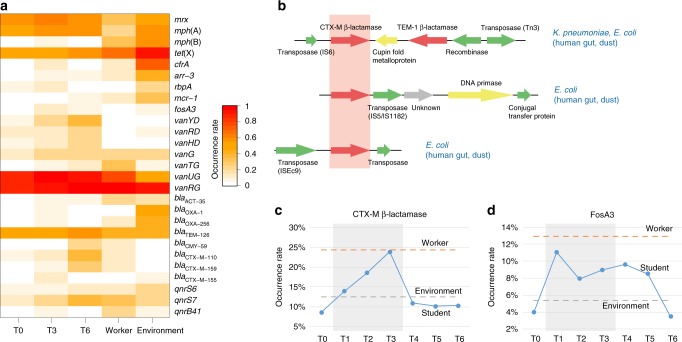
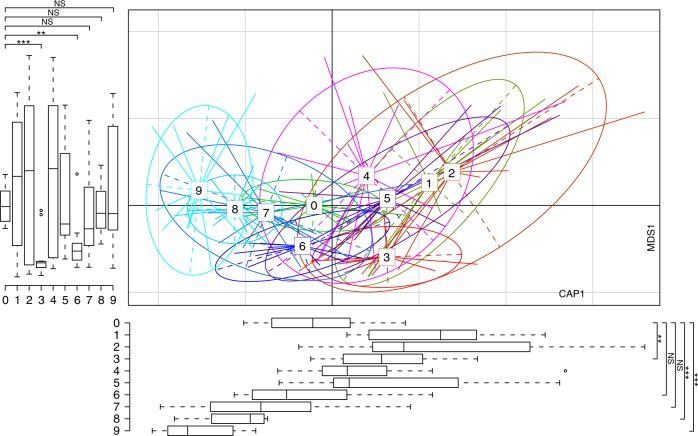
Anthropogenic environments have been implicated in enrichment and exchange of antibiotic resistance genes and bacteria. Here we study the impact of confined and controlled swine farm environments on temporal changes in the gut microbiome and resistome of veterinary students with occupational exposure for 3 months. By analyzing 16S rRNA and whole metagenome shotgun sequencing data in tandem with culture-based methods, we show that farm exposure shapes the gut microbiome of students, resulting in enrichment of potentially pathogenic taxa and antimicrobial resistance genes. Comparison of students' gut microbiomes and resistomes to farm workers' and environmental samples revealed extensive sharing of resistance genes and bacteria following exposure and after three months of their visit. Notably, antibiotic resistance genes were found in similar genetic contexts in student samples and farm environmental samples. Dynamic Bayesian network modeling predicted that the observed changes partially reverse over a 4-6 month period. Our results indicate that acute changes in a human's living environment can persistently shape their gut microbiota and antibiotic resistome.
Conflict of interest statement
The authors declare no competing interests.
Figures