

Oncogene-independent BCR-like signaling adaptation confers drug resistance in Ph-like ALL
- PMID: 32191635
- PMCID: PMC7324172
- DOI: 10.1172/JCI134424
Oncogene-independent BCR-like signaling adaptation confers drug resistance in Ph-like ALL
Abstract
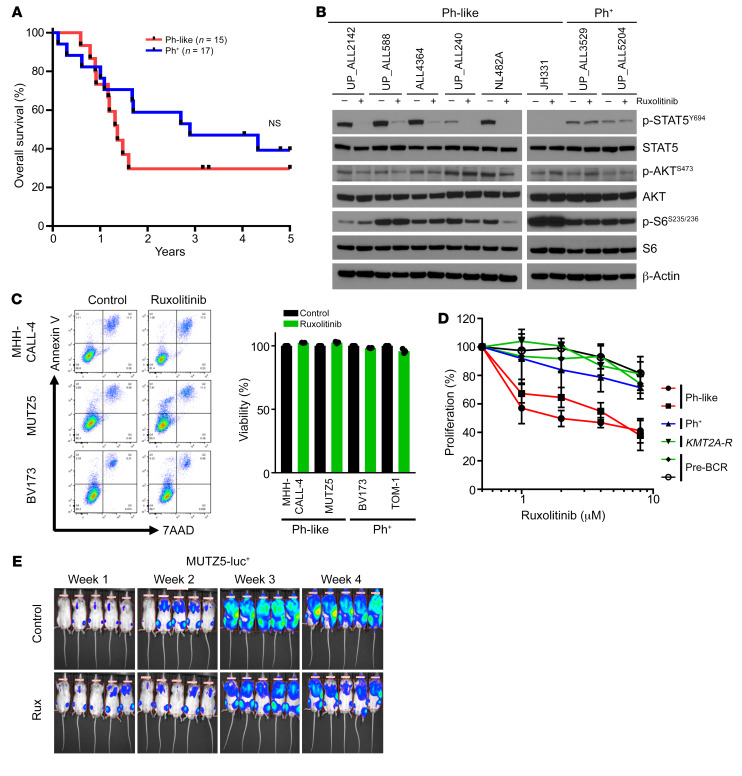
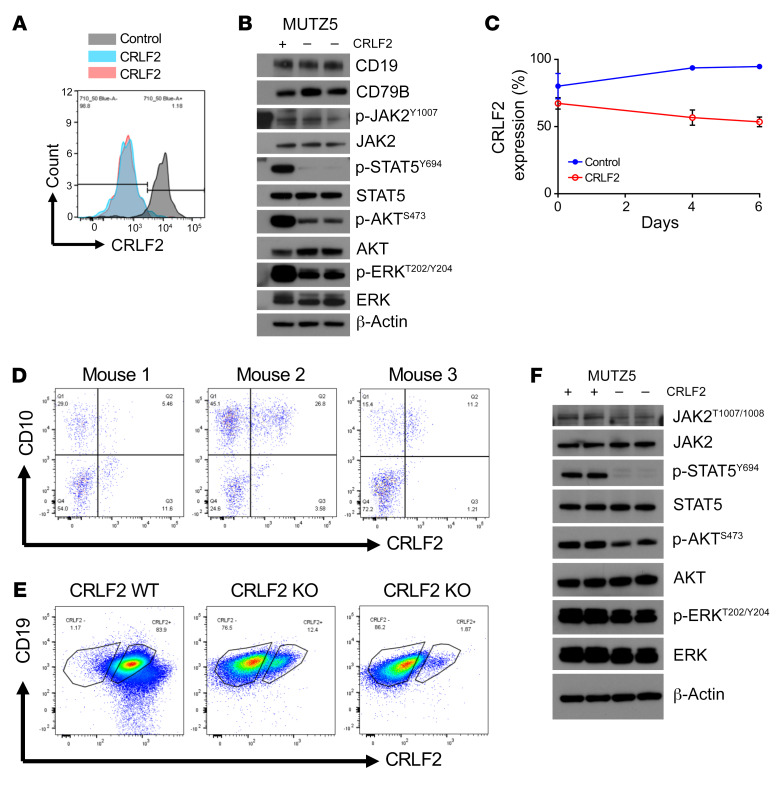
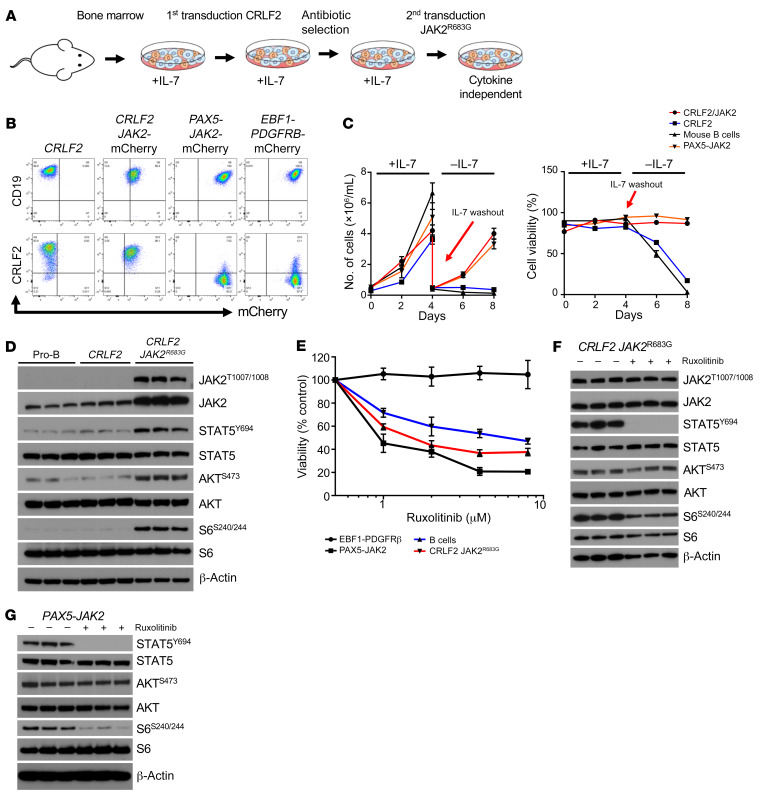
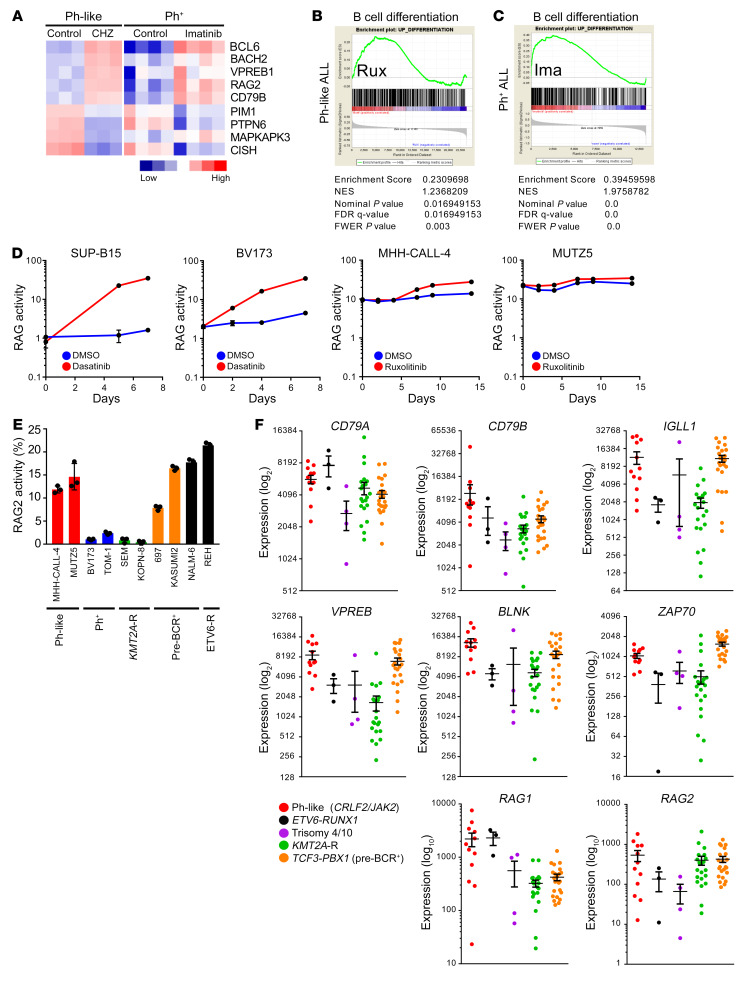
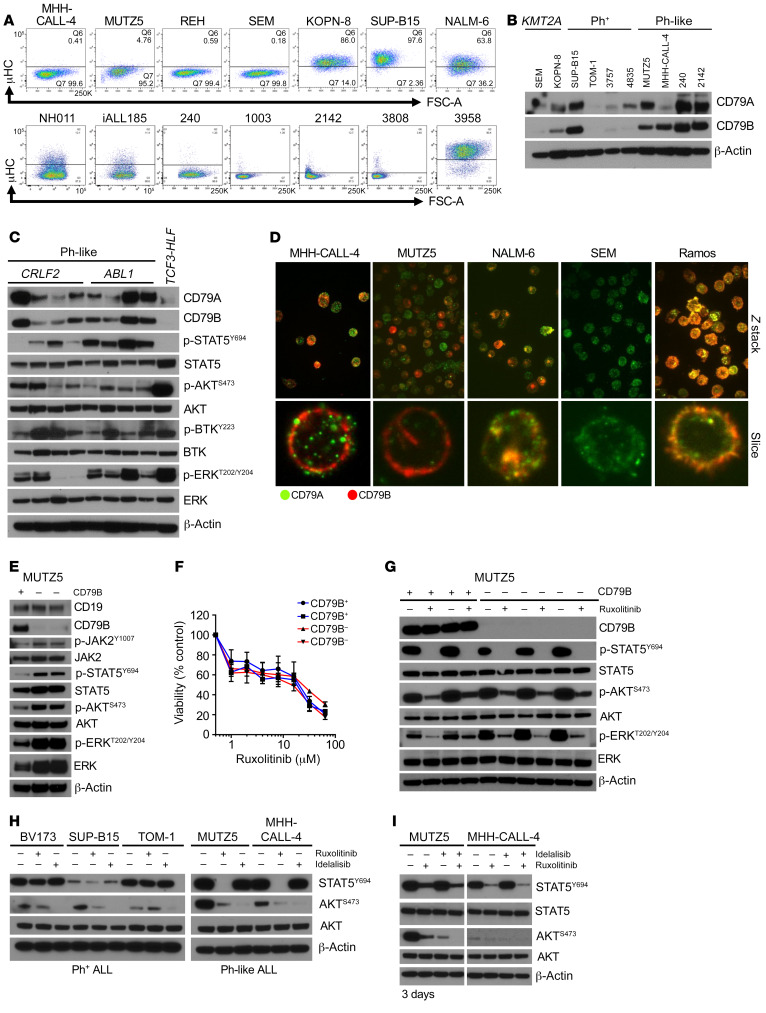
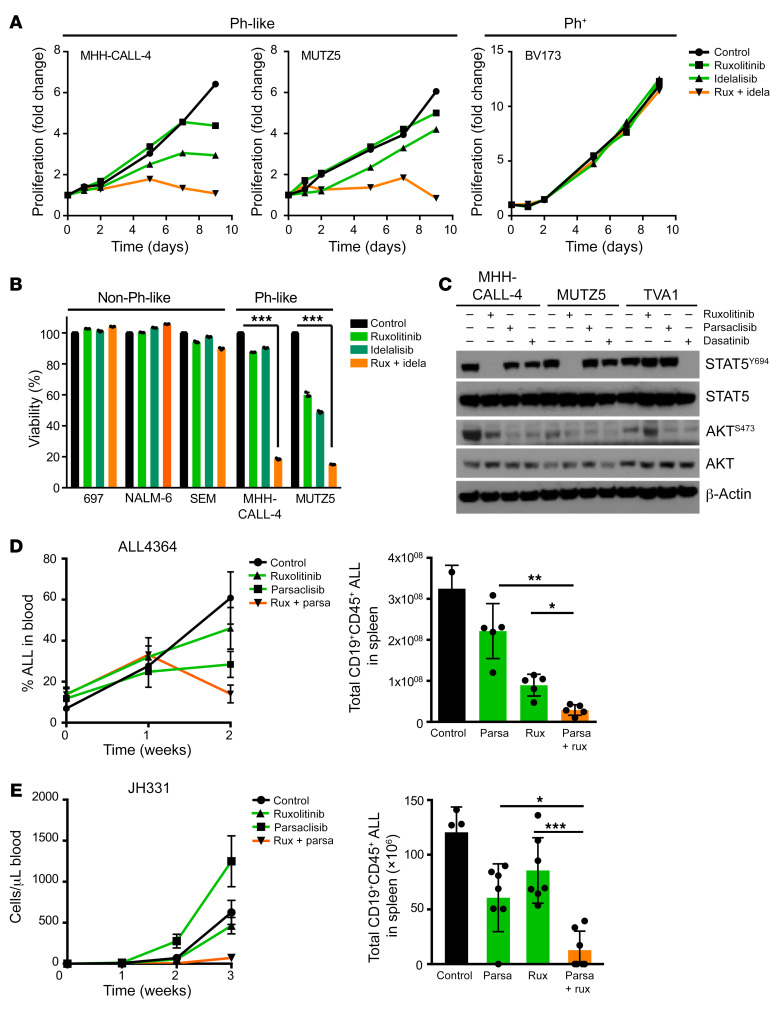
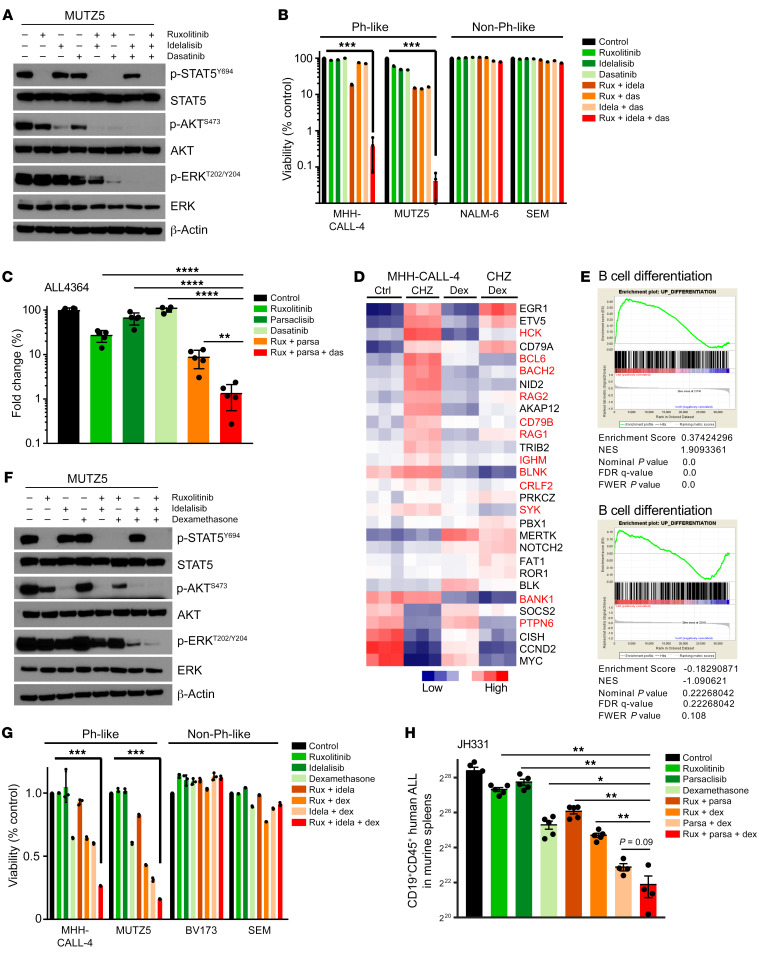
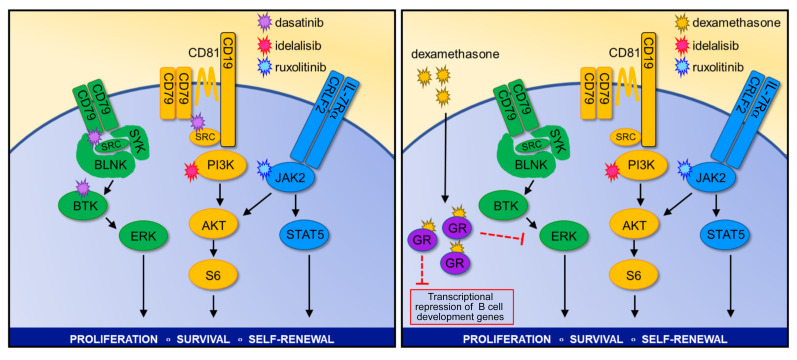
Children and adults with Philadelphia chromosome-like B cell acute lymphoblastic leukemia (Ph-like B-ALL) experience high relapse rates despite best-available conventional chemotherapy. Ph-like ALL is driven by genetic alterations that activate constitutive cytokine receptor and kinase signaling, and early-phase trials are investigating the potential of the addition of tyrosine kinase inhibitors (TKIs) to chemotherapy to improve clinical outcomes. However, preclinical studies have shown that JAK or PI3K pathway inhibition is insufficient to eradicate the most common cytokine receptor-like factor 2-rearranged (CRLF2-rearranged) Ph-like ALL subset. We thus sought to define additional essential signaling pathways required in Ph-like leukemogenesis for improved therapeutic targeting. Herein, we describe an adaptive signaling plasticity of CRLF2-rearranged Ph-like ALL following selective TKI pressure, which occurs in the absence of genetic mutations. Interestingly, we observed that Ph-like ALL cells have activated SRC, ERK, and PI3K signaling consistent with activated B cell receptor (BCR) signaling, although they do not express cell surface μ-heavy chain (μHC). Combinatorial targeting of JAK/STAT, PI3K, and "BCR-like" signaling with multiple TKIs and/or dexamethasone prevented this signaling plasticity and induced complete cell death, demonstrating a more optimal and clinically pragmatic therapeutic strategy for CRLF2-rearranged Ph-like ALL.
Keywords: Hematology; Leukemias; Oncology; Protein kinases; Signal transduction.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
