

PPanGGOLiN: Depicting microbial diversity via a partitioned pangenome graph
- PMID: 32191703
- PMCID: PMC7108747
- DOI: 10.1371/journal.pcbi.1007732
PPanGGOLiN: Depicting microbial diversity via a partitioned pangenome graph
Erratum in
-
Correction: PPanGGOLiN: Depicting microbial diversity via a partitioned pangenome graph.PLoS Comput Biol. 2021 Dec 10;17(12):e1009687. doi: 10.1371/journal.pcbi.1009687. eCollection 2021 Dec. PLoS Comput Biol. 2021. PMID: 34890406 Free PMC article.
Abstract
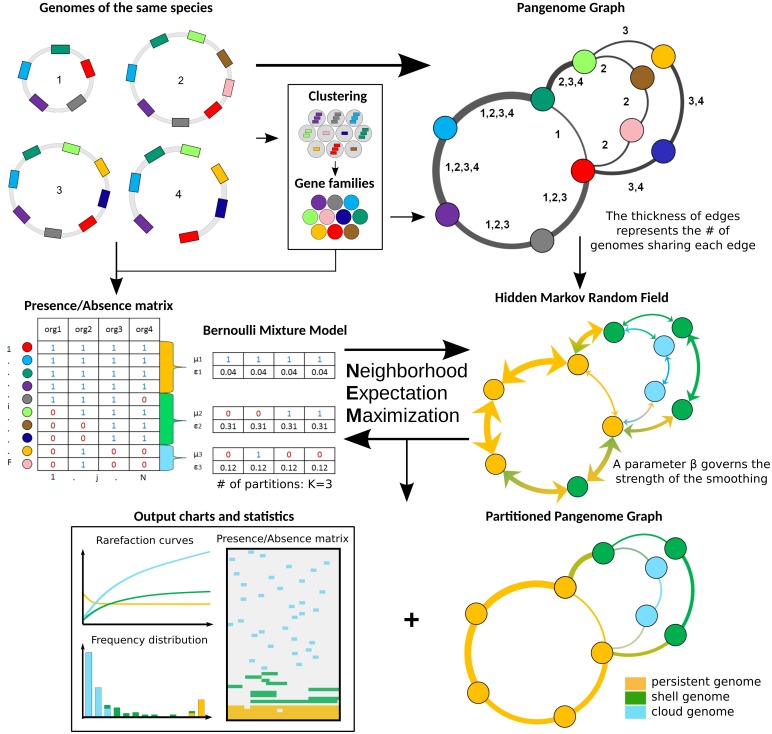
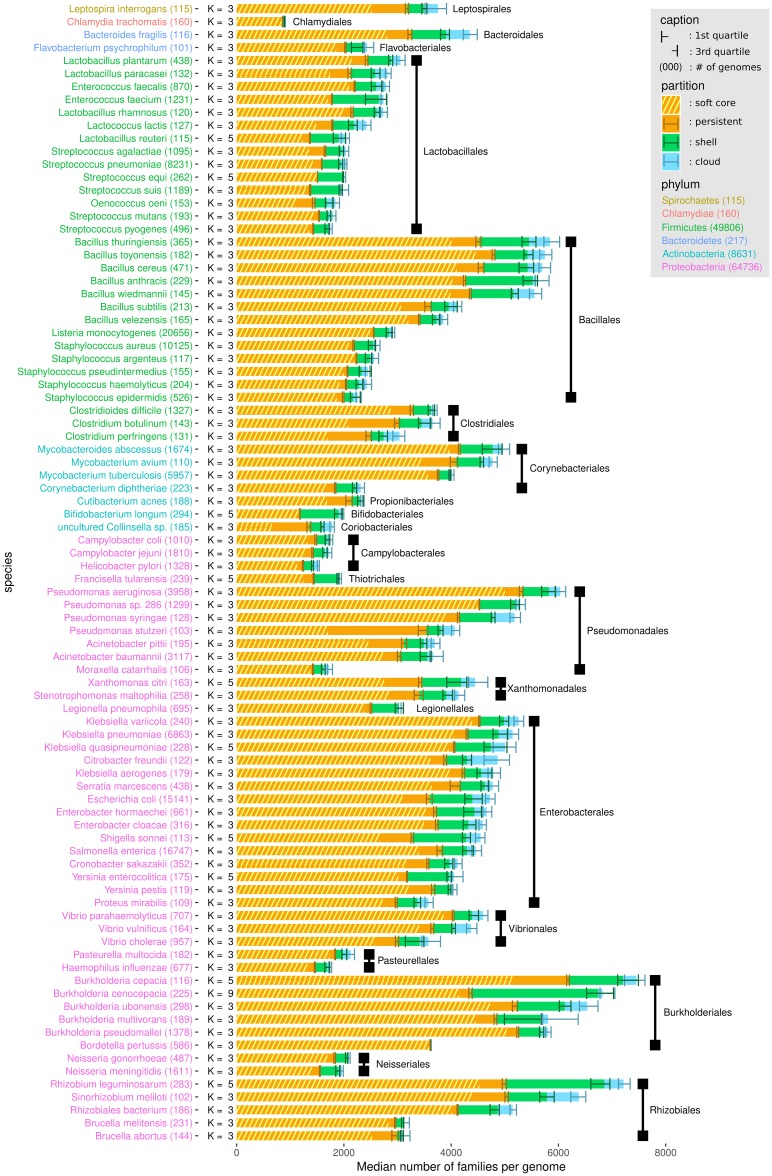
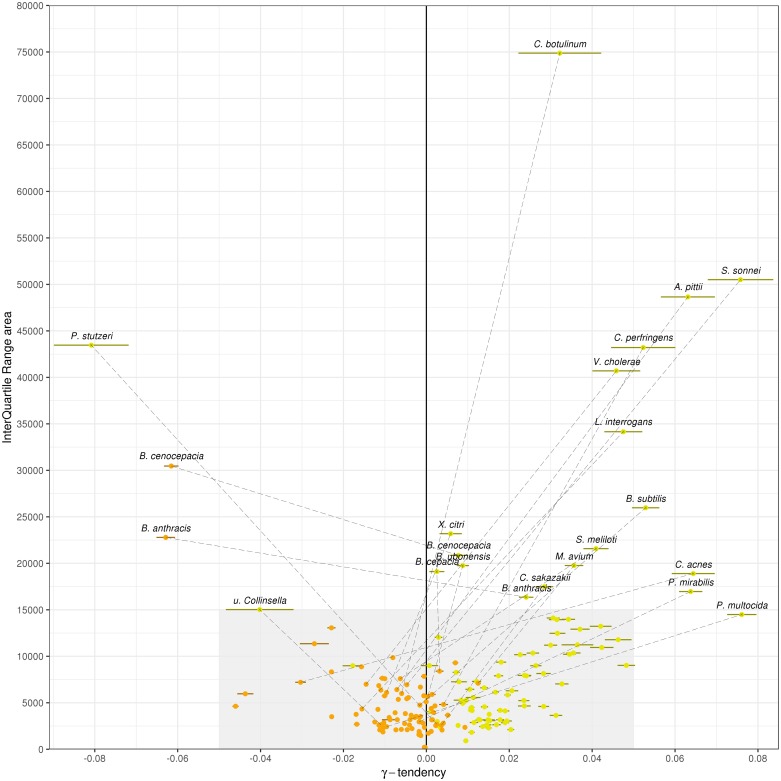
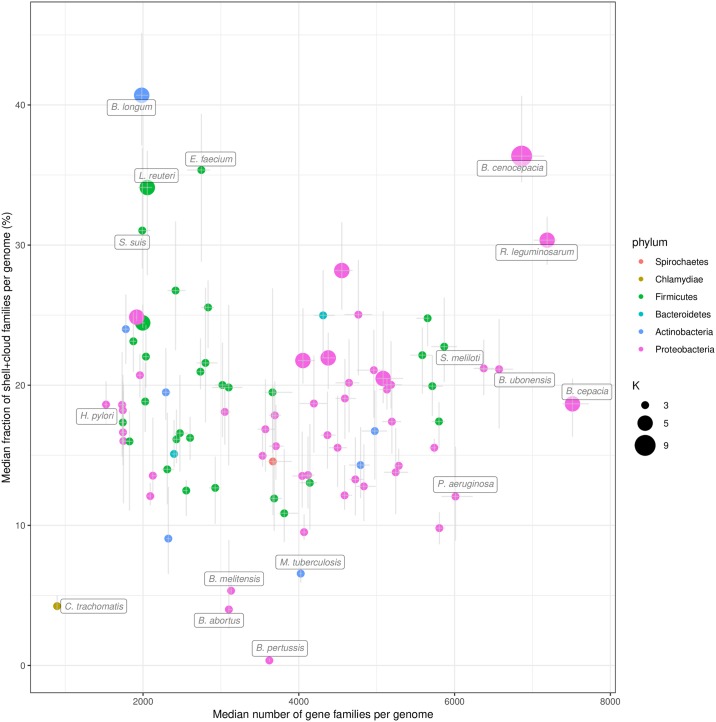
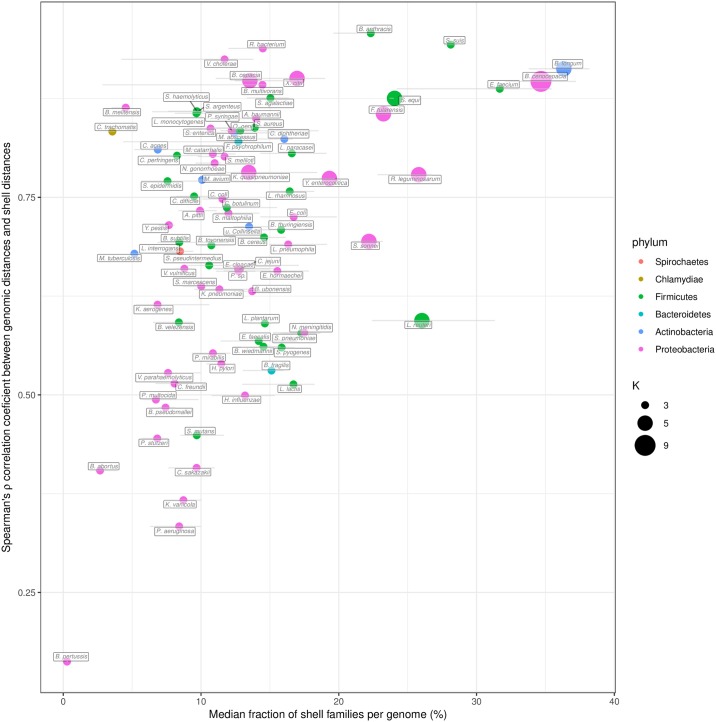
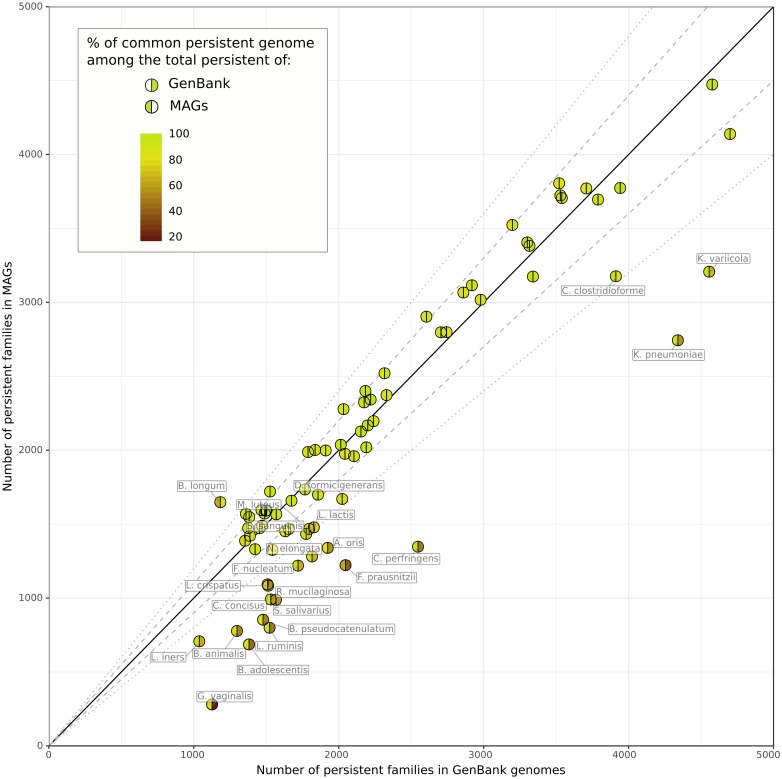
The use of comparative genomics for functional, evolutionary, and epidemiological studies requires methods to classify gene families in terms of occurrence in a given species. These methods usually lack multivariate statistical models to infer the partitions and the optimal number of classes and don't account for genome organization. We introduce a graph structure to model pangenomes in which nodes represent gene families and edges represent genomic neighborhood. Our method, named PPanGGOLiN, partitions nodes using an Expectation-Maximization algorithm based on multivariate Bernoulli Mixture Model coupled with a Markov Random Field. This approach takes into account the topology of the graph and the presence/absence of genes in pangenomes to classify gene families into persistent, cloud, and one or several shell partitions. By analyzing the partitioned pangenome graphs of isolate genomes from 439 species and metagenome-assembled genomes from 78 species, we demonstrate that our method is effective in estimating the persistent genome. Interestingly, it shows that the shell genome is a key element to understand genome dynamics, presumably because it reflects how genes present at intermediate frequencies drive adaptation of species, and its proportion in genomes is independent of genome size. The graph-based approach proposed by PPanGGOLiN is useful to depict the overall genomic diversity of thousands of strains in a compact structure and provides an effective basis for very large scale comparative genomics. The software is freely available at https://github.com/labgem/PPanGGOLiN.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
