

Drug Sensitivity and Allele Specificity of First-Line Osimertinib Resistance EGFR Mutations
- PMID: 32193290
- PMCID: PMC7392201
- DOI: 10.1158/0008-5472.CAN-19-3819
Drug Sensitivity and Allele Specificity of First-Line Osimertinib Resistance EGFR Mutations
Abstract
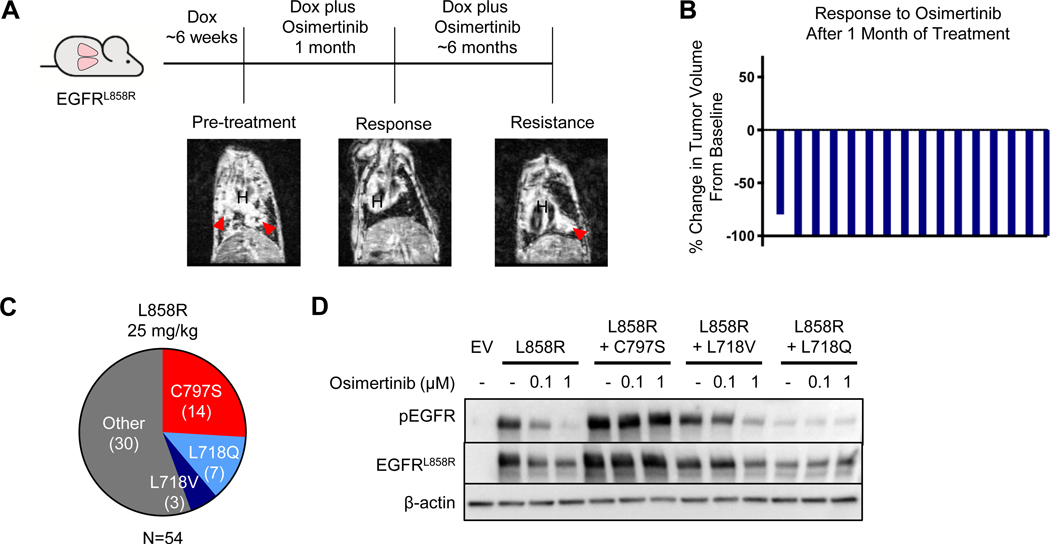
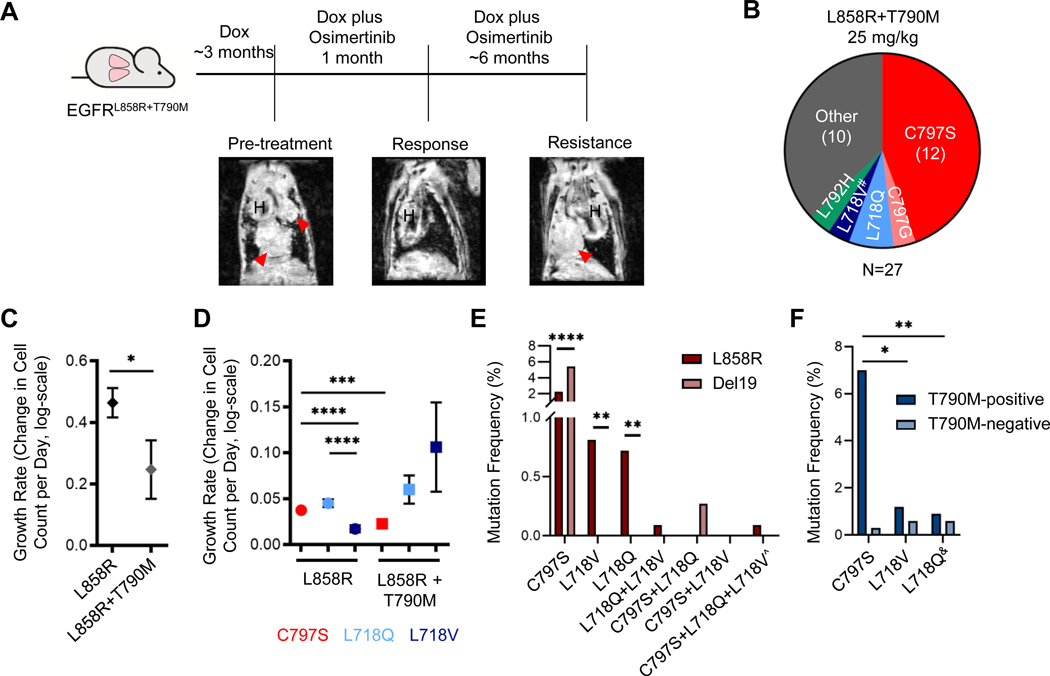
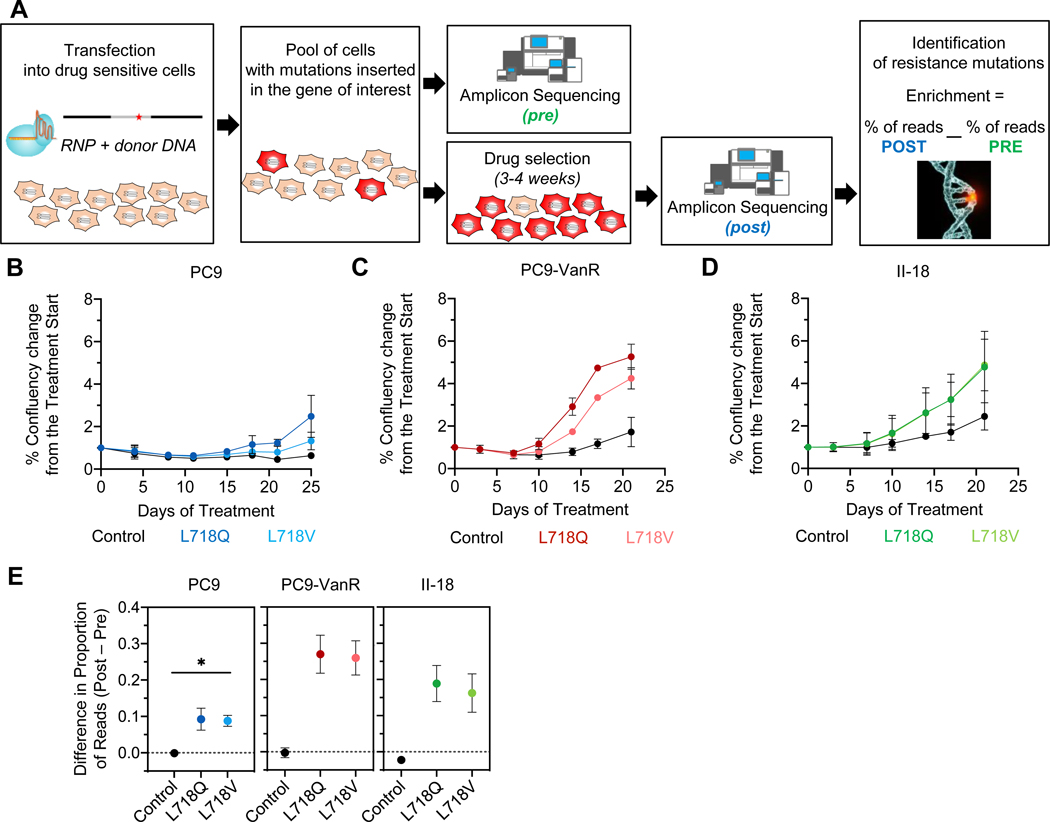
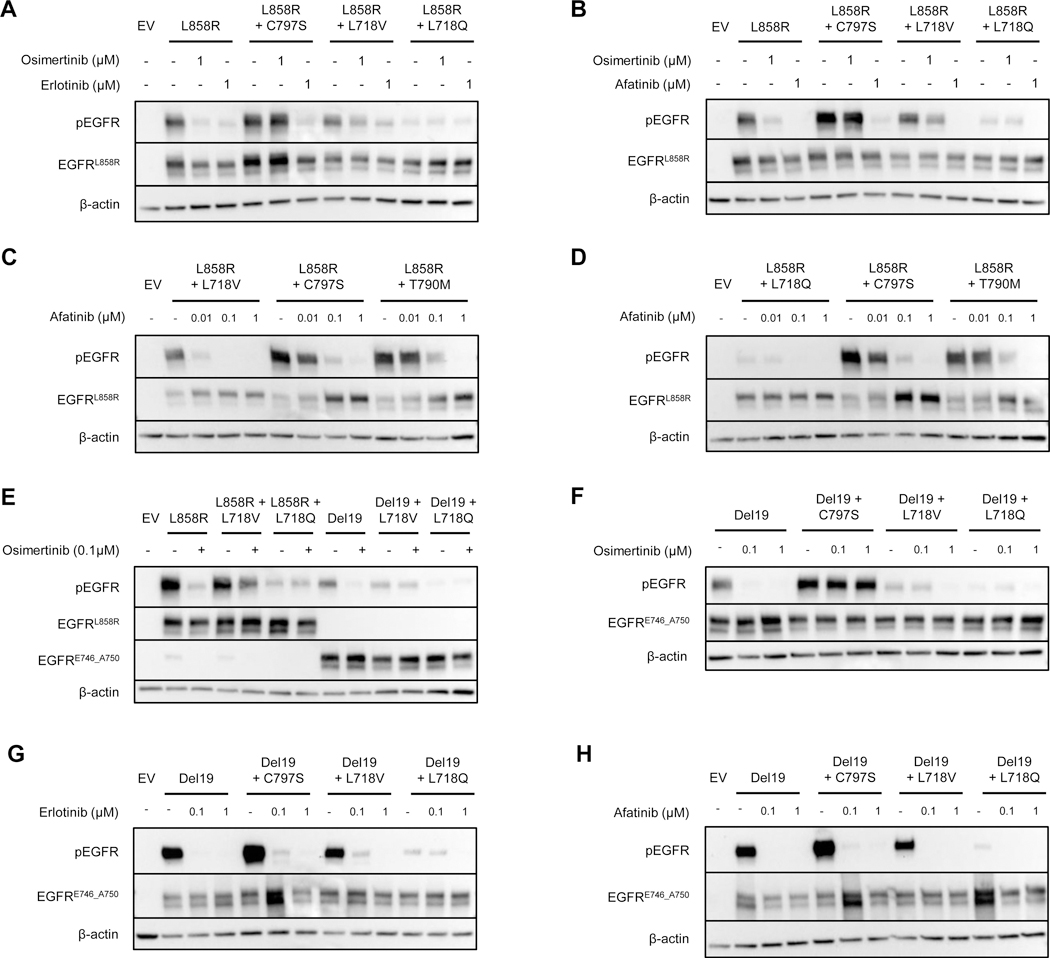
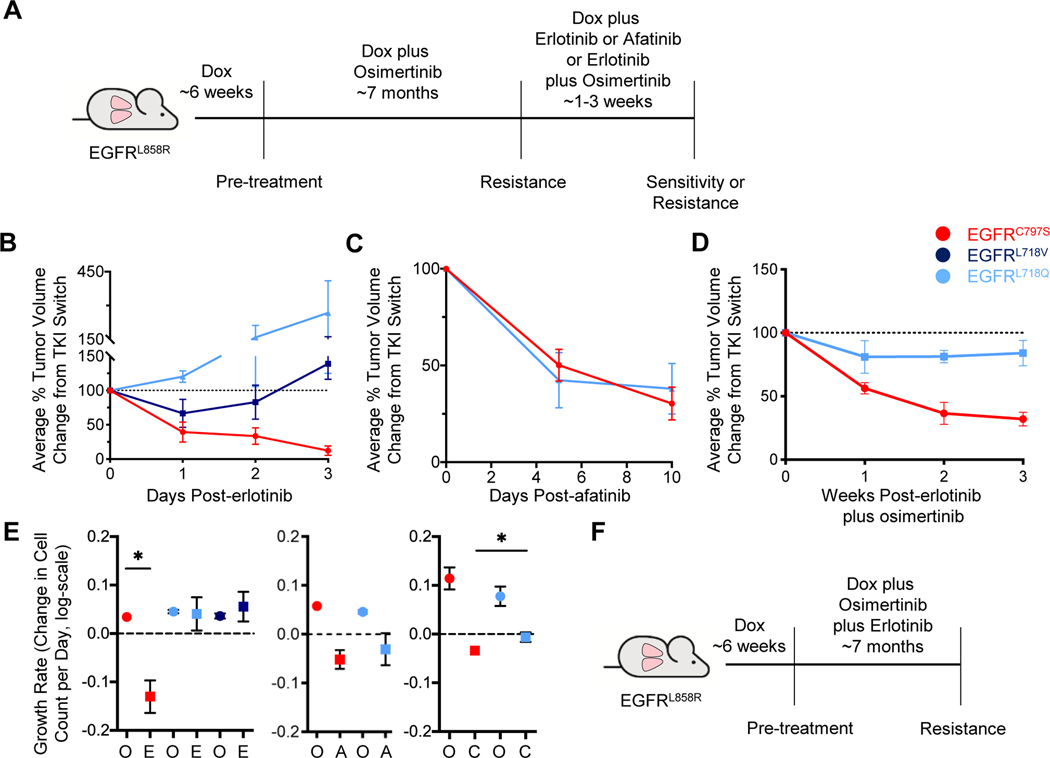
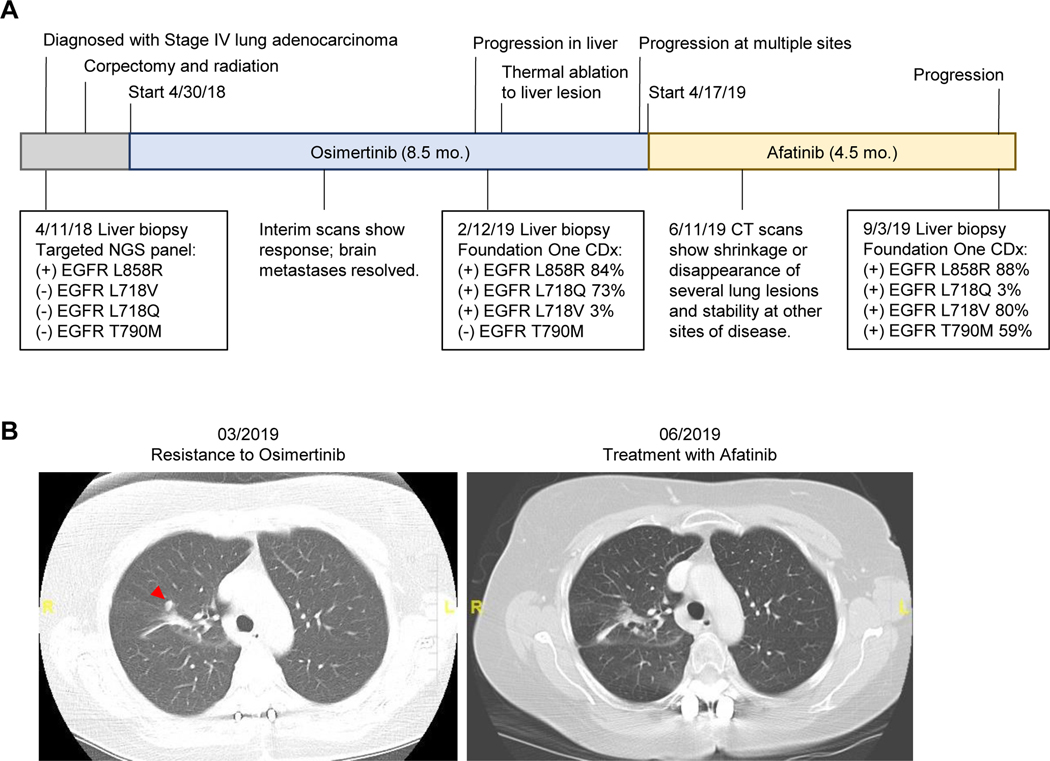
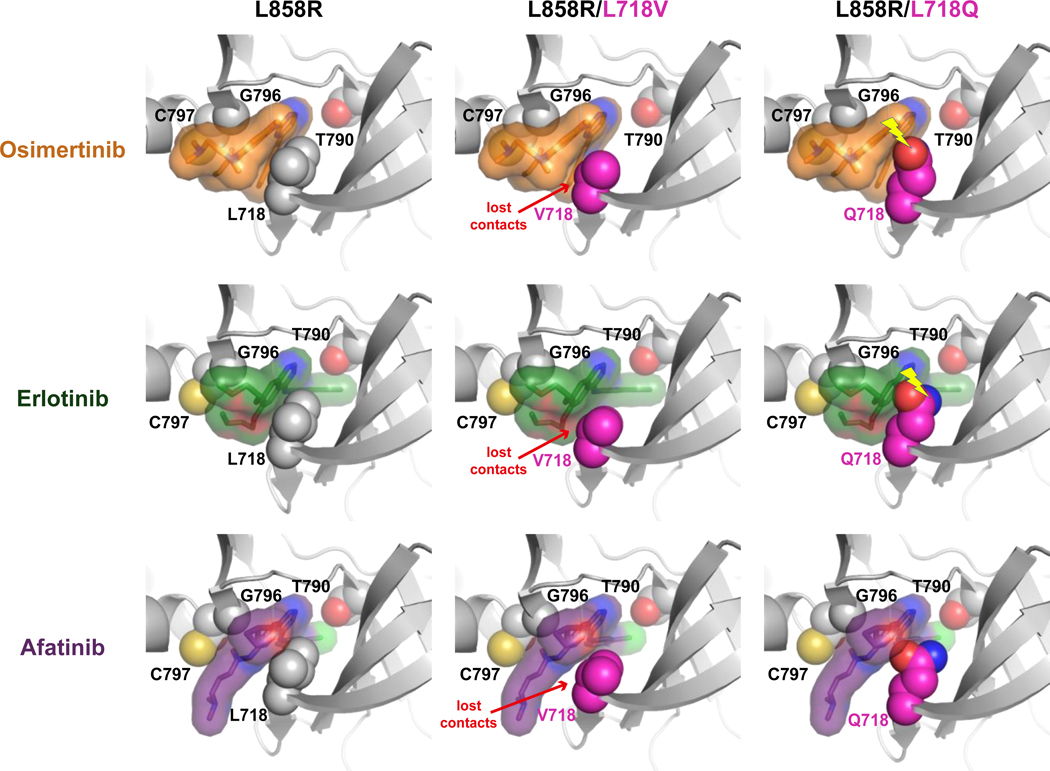
Osimertinib, a mutant-specific third-generation EGFR tyrosine kinase inhibitor, is emerging as the preferred first-line therapy for EGFR-mutant lung cancer, yet resistance inevitably develops in patients. We modeled acquired resistance to osimertinib in transgenic mouse models of EGFRL858R -induced lung adenocarcinoma and found that it is mediated largely through secondary mutations in EGFR-either C797S or L718V/Q. Analysis of circulating free DNA data from patients revealed that L718Q/V mutations almost always occur in the context of an L858R driver mutation. Therapeutic testing in mice revealed that both erlotinib and afatinib caused regression of osimertinib-resistant C797S-containing tumors, whereas only afatinib was effective on L718Q mutant tumors. Combination first-line osimertinib plus erlotinib treatment prevented the emergence of secondary mutations in EGFR. These findings highlight how knowledge of the specific characteristics of resistance mutations is important for determining potential subsequent treatment approaches and suggest strategies to overcome or prevent osimertinib resistance in vivo. SIGNIFICANCE: This study provides insight into the biological and molecular properties of osimertinib resistance EGFR mutations and evaluates therapeutic strategies to overcome resistance. GRAPHICAL ABSTRACT: http://cancerres.aacrjournals.org/content/canres/80/10/2017/F1.large.jpg.
©2020 American Association for Cancer Research.
Conflict of interest statement
Conflict of Interest Statement
JHS, DA, MG, HK, TFS, AK, RH, FM, K.Poels, IKvR, MAL, AN, DF, KDA, WWL and AU do not declare any conflicts of interest.
Figures
References
-
- Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. 2014;11(8):473–81. - PubMed
-
- Janne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015;372(18):1689–99. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
