

Insights into human genetic variation and population history from 929 diverse genomes
- PMID: 32193295
- PMCID: PMC7115999
- DOI: 10.1126/science.aay5012
Insights into human genetic variation and population history from 929 diverse genomes
Abstract
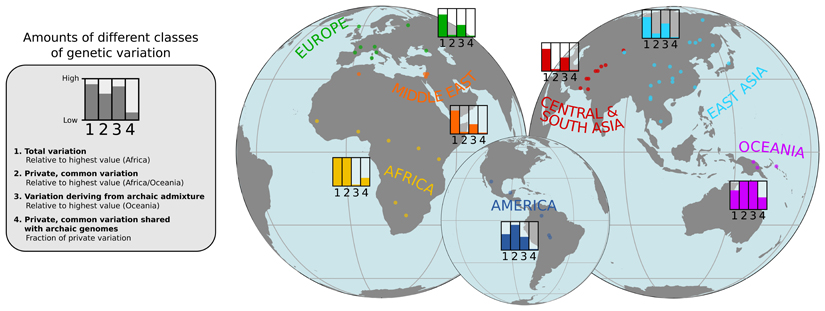
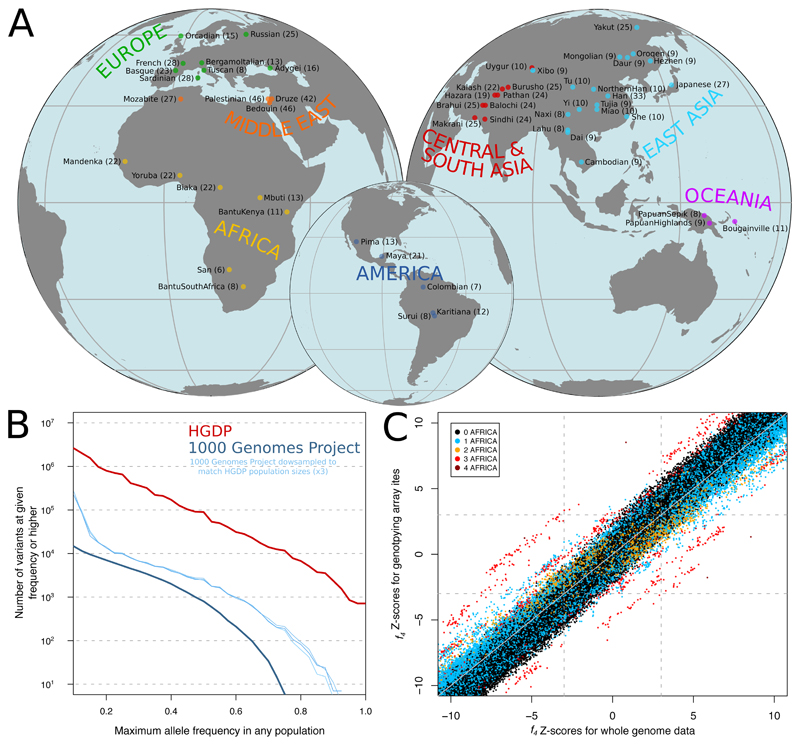
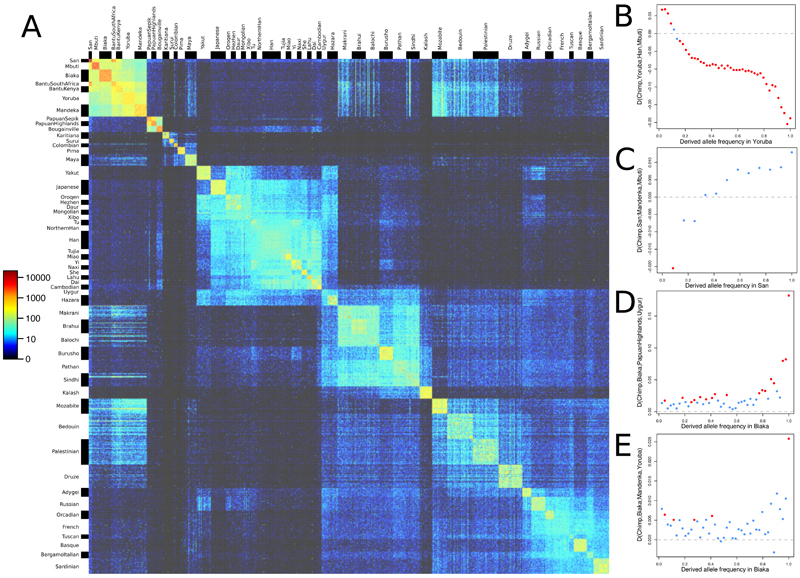
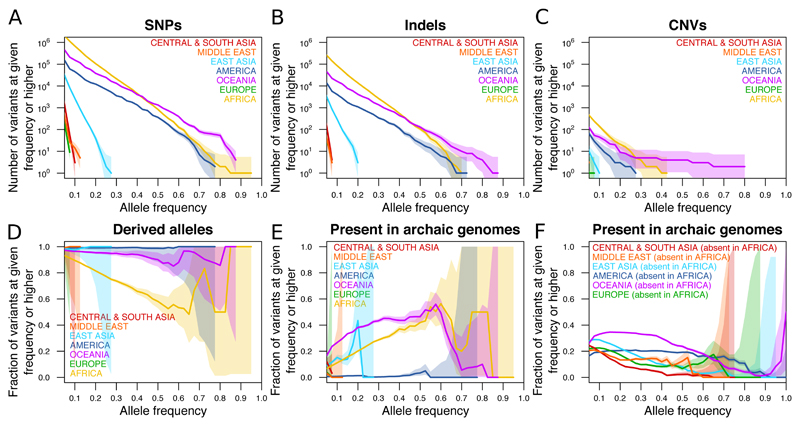
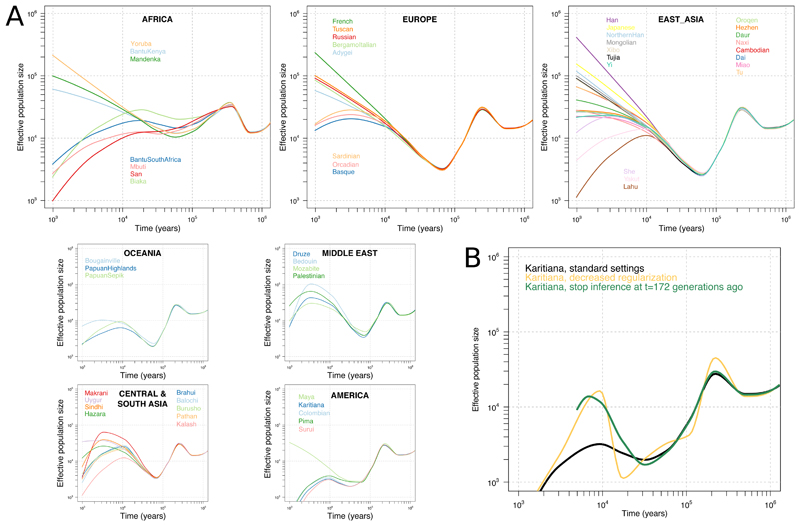
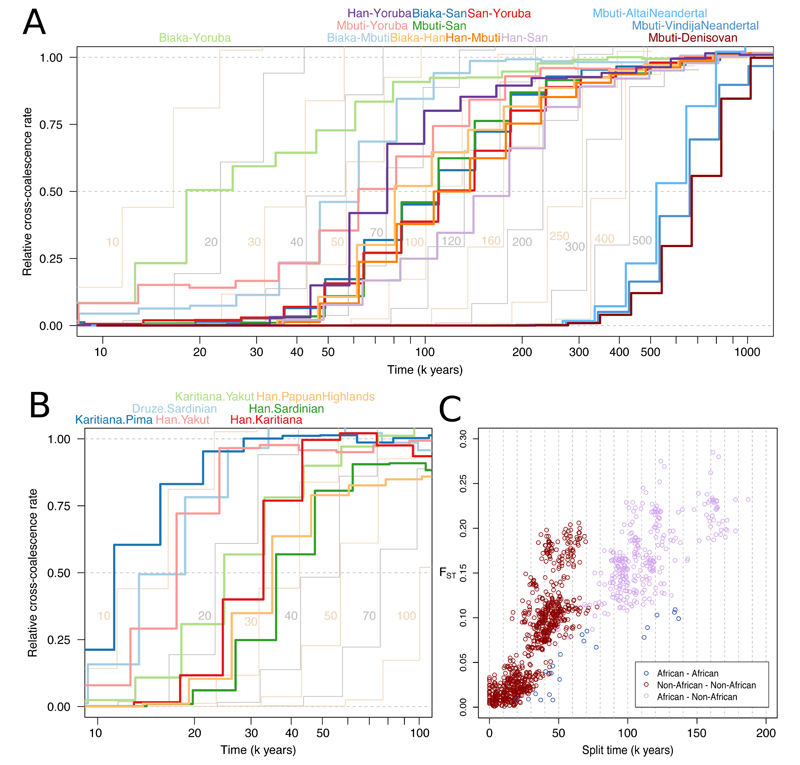
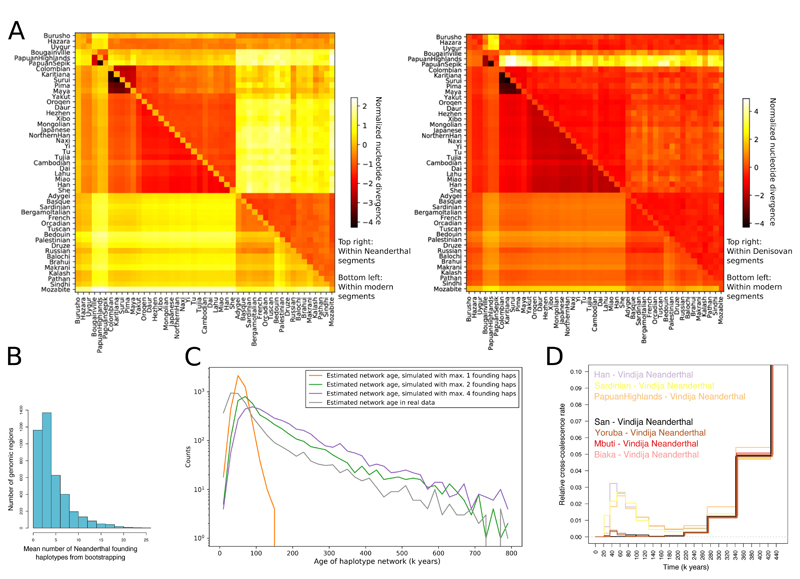
Genome sequences from diverse human groups are needed to understand the structure of genetic variation in our species and the history of, and relationships between, different populations. We present 929 high-coverage genome sequences from 54 diverse human populations, 26 of which are physically phased using linked-read sequencing. Analyses of these genomes reveal an excess of previously undocumented common genetic variation private to southern Africa, central Africa, Oceania, and the Americas, but an absence of such variants fixed between major geographical regions. We also find deep and gradual population separations within Africa, contrasting population size histories between hunter-gatherer and agriculturalist groups in the past 10,000 years, and a contrast between single Neanderthal but multiple Denisovan source populations contributing to present-day human populations.
Copyright © 2020 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Figures
Comment in
-
Diverse human genomes.Nat Rev Genet. 2020 Jun;21(6):338. doi: 10.1038/s41576-020-0235-y. Nat Rev Genet. 2020. PMID: 32269330 No abstract available.
References
-
- Cann HM, et al. A human genome diversity cell line panel. Science. 2002;296:261–262. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
