

webGQT: A Shiny Server for Genotype Query Tools for Model-Based Variant Filtering
- PMID: 32194629
- PMCID: PMC7063093
- DOI: 10.3389/fgene.2020.00152
webGQT: A Shiny Server for Genotype Query Tools for Model-Based Variant Filtering
Abstract
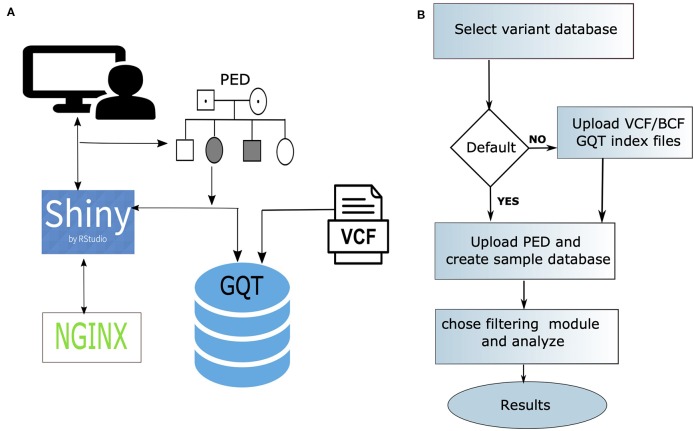
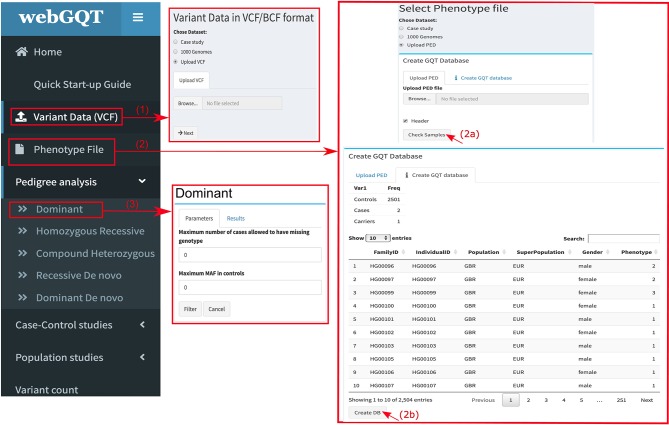
Summary: Genotype Query Tools (GQT) were developed to discover disease-causing variations from billions of genotypes and millions of genomes, processes data at substantially higher speed over other existing methods. While GQT has been available to a wide audience as command-line software, the difficulty of constructing queries among non-IT or non-bioinformatics researchers has limited its applicability. To overcome this limitation, we developed webGQT, an easy-to-use tool with a graphical user interface. With pre-built queries across three modules, webGQT allows for pedigree analysis, case-control studies, and population frequency studies. As a package, webGQT allows researchers with less or no applied bioinformatics/IT experience to mine potential disease-causing variants from billions.
Results: webGQT offers a flexible and easy-to-use interface for model-based candidate variant filtering for Mendelian diseases from thousands to millions of genomes at a reduced computation time. Additionally, webGQT provides adjustable parameters to reduce false positives and rescue missing genotypes across all modules. Using a case study, we demonstrate the applicability of webGQT to query non-human genomes. In addition, we demonstrate the scalability of webGQT on large data sets by implementing complex population-specific queries on the 1000 Genomes Project Phase 3 data set, which includes 8.4 billion variants from 2504 individuals across 26 different populations. Furthermore, webGQT supports filtering single-nucleotide variants, short insertions/deletions, copy number or any other variant genotypes supported by the VCF specification. Our results show that webGQT can be used as an online web service, or deployed on personal computers or local servers within research groups.
Availability: webGQT is made available to the users in three forms: 1) as a webserver available at https://vm1138.kaj.pouta.csc.fi/webgqt/, 2) as an R package to install on personal computers, and 3) as part of the same R package to configure on the user's own servers. The application is available for installation at https://github.com/arumds/webgqt.
Keywords: Bigdata; GQT; R package; filtering; shiny server; variant; webGQT.
Copyright © 2020 Arumilli, Layer, Hytönen and Lohi.
Figures
References
-
- Cingolani P., Platts A., Wang Le L., Coon M., Nguyen T., Wang L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92. 10.4161/fly.19695 - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials